

Партнерка на США и Канаду по недвижимости, выплаты в крипто
- 30% recurring commission
- Выплаты в USDT
- Вывод каждую неделю
- Комиссия до 5 лет за каждого referral
На правах рукописи
САВВАТЕЕВ АЛЕКСЕЙ МИХАЙЛОВИЧ
ОЦЕНКА КАЧЕСТВА И СТАНДАРТИЗАЦИЯ
КОМПОЗИЦИЙ НА БАЗЕ ДИКВЕРТИНА
15.00.02 – фармацевтическая химия, фармакогнозия
АВТОРЕФЕРАТ
диссертации на соискание ученой степени
кандидата фармацевтических наук
МОСКВА – 2007
Работа выполнена в Государственном образовательном учреждении высшего профессионального образования Московская медицинская академия имени Федерального агентства по здравоохранению и социальному развитию
Научный руководитель:
доктор фармацевтических наук
Официальные оппоненты:
профессор, доктор химических наук
профессор, доктор фармацевтических наук
Ведущая организация:
Институт стандартизации и контроля лекарственных средств ФГУ «Научный центр экспертизы средств медицинского применения» Росздравнадзора
Защита состоится «____»_______________2007 г. в _____ часов на заседании диссертационного совета Д.208.040.09 при Московской медицинской академии имени г. Москва, Никитский бульвар, 13.
С диссертацией можно ознакомиться в библиотеке Московской медицинской академии имени г. Москва, Нахимовский проспект, 49.
Автореферат разослан «____» __________2007 г.
Ученый секретарь
диссертационного совета Д.208.040.09
доктор фармацевтических наук, профессор Садчикова Наталья
Петровна
ОБЩАЯ ХАРАКТЕРИСТИКА РАБОТЫ
Актуальность темы. Для России с ее богатейшими растительными ресурсами характерен широкий масштаб производства фитопрепаратов. В состав многих из них входят флавоноидные соединения.
Относительно недавно этот список пополнился препаратом Диквертин®, обладающим антиоксидантными и капилляропротекторными свойствами. Растительным источником диквертина служит древесина лиственницы. Состав его представлен тремя флавоноидными соединениями при подавляющем количестве (90% и выше) одного из них – дигидрокверцетина.
Банк присущих диквертину фундаментальных фармакологических свойств обеспечивает ему базисный статус для создания на его основе препаратов, комбинированных с другими лекарственными средствами с целью оптимизации отдельных видов фармакологического действия. В последние годы проведены совместные исследования сотрудниками НИИ фармакологии ТНЦ СО РАМН и кафедры органической химии ММА им. по разработке принципиально новой группы гемореологических лекарственных препаратов. Известно, что гемореологические нарушения осложняют течение таких заболеваний, как ишемическая болезнь сердца, артериальная гипертензия и др.
Препарат, сочетающий диквертин и аскорбиновую кислоту, зарегистрирован как ангиопротекторное лекарственное средство Асковертин®. Препарат, сочетающий диквертин и ацетилсалициловую кислоту (под рабочим названием Саливертин), прошел стадию доклинического изучения с выявлением антитромбоцитарного и гемореологического действия. Cочетание диквертина с двумя компонентами – аскорбиновой кислотой и β-каротином – используется как биологически активная добавка к пище «Каровертин».
Появление новых комбинированных препаратов в силу усложнения состава действующих веществ, существенно различающихся по свойствам, ставит задачу поиска оптимальных путей их фармацевтического анализа.
Актуальность настоящей работы определяется необходимостью совершенствования на современном физико-химическом уровне стандартизации базового фитопрепарата диквертина, а также создания системы стандартизации новых комбинированных препаратов.
Цель и задачи исследования. Целью настоящего исследования являлась разработка методологического подхода к оптимизации хроматографического анализа в качестве сквозного унифицированного метода стандартизации как базового препарата диквертина, так и комбинированных препаратов на его основе.
Для достижения поставленной цели решались следующие задачи:
· модификация и унификация методик анализа диквертина – от растительного сырья, субстанции, стандартного образца до лекарственной формы;
· совершенствование методик спектрального (УФ, ИК, ЯМР) анализа стандартного образца дигидрокверцетина;
· разработка методик хроматографического анализа с учетом специфичности состава комбинированных препаратов – асковертина, саливертина и БАД – каровертина;
· валидация предлагаемых методик на основе изучения соответствующих валидационных характеристик;
· включение разработанных методик в фармакопейную документацию.
Научная новизна. Предложен единый методический подход к стандартизации комбинированных препаратов на базе диквертина путем сквозного использования метода ВЭЖХ в сочетании со специфичностью пробоподготовки.
Определены оптимальные хроматографические условия эффективного разделения флавоноидных компонентов диквертина и аскорбиновой кислоты, компонентов диквертина и ацетилсалициловой и салициловой кислот в составе комбинированных препаратов – асковертина и саливертина соответственно. Разработаны селективные и воспроизводимые методики качественного и количественного анализа действующих веществ комбинированных препаратов.
Предложены спектральные (УФ-, ИК-, ЯМР 1Н, ЯМР 13С) и хроматографические критерии подлинности и оценки качества стандартного образца дигидрокверцетина.
Разработан оригинальный комбинированный способ использования в определенной последовательности методов твердофазной экстракции, ВЭЖХ и спектрофотометрии для анализа разнополярных компонентов каровертина.
Впервые разработана система стандартизации лекарственного растительного сырья – древесины лиственницы цельной и измельченной.
Практическая значимость. Методики анализа действующих веществ препаратов диквертина, асковертина и саливертина валидированы и унифицированы с целью определения таких фармакопейных показателей, как подлинность, родственные соединения, посторонние примеси, растворение, однородность дозирования, количественное определение. Разработанные аналитические методики включены в зарегистрированные фармакопейные статьи: Лиственницы древесина цельная, измельченная «ангро»; Дигидрокверцетин–стандартный образец; Диквертин субстанция; Диквертин таблетки; Асковертин таблетки; в проект фармакопейной статьи Саливертин таблетки; в Технические условия на БАД «Каровертин».
Предложенные методики апробированы на производственных сериях таблеток диквертина, асковертина и саливертина.
Основные положения, выносимые на защиту.
· Результаты изучения хроматографических характеристик флавоноидных компонентов диквертина. Усовершенствованная методика стандартизации и оценки качества диквертина методом ВЭЖХ.
· Спектральная (УФ-, ИК-, ЯМР 1Н и ЯМР 13С) и хроматографическая характеристика дигидрокверцетина как стандартного образца и оценка его индивидуальности и степени чистоты.
· Оптимальные хроматографические параметры ВЭЖХ для одновременного качественного и количественного анализа аскорбиновой кислоты и флавоноидов диквертина. Валидированная и унифицированная для определения ряда фармакопейных показателей методика стандартизации препарата асковертина методом ВЭЖХ.
· Оптимальные хроматографические параметры ВЭЖХ для одновременного качественного и количественного анализа ацетилсалициловой кислоты, примеси салициловой кислоты и флавоноидов диквертина. Валидированная и унифицированная для определения фармакопейных показателей методика стандартизации желудочно-резистентных таблеток саливертина методом ВЭЖХ.
· Комплексный аналитический подход (твердофазная экстракция, ВЭЖХ, спектрофотометрия) к анализу компонентов каровертина (аскорбиновая кислота, диквертин, b-каротин).
· Стандартизация лекарственного растительного сырья – древесины лиственницы цельной, измельченной «ангро».
· Результаты изучения возможности использования разработанных методик в анализе опытно-промышленных серий асковертина и лабораторных серий саливертина.
Апробация работы. Основные положения работы доложены на IV, VI, IX и X международных съездах «Актуальные проблемы создания новых лекарственных препаратов природного происхождения» (Великий Новгород 2001, С-Пб 2002, 2005 и 2006), на VI Симпозиуме по фенольным соединениям (Москва 2004) на XII Российском национальном конгрессе «Человек и лекарство» (Москва 2005).
Публикации. По теме диссертации опубликовано 9 печатных работ
Связь задач исследования с проблемным планом фармацевтических наук. Диссертационная работа выполнена в рамках комплексной темы кафедры органической химии ММА им. : «Физико-химические основы стандартизации и биотрансформации лекарственных средств и биологически активных добавок к пище». Номер госрегистрации .
Объем и структура диссертации. Диссертация изложена на 0000 страницах машинописного текста и состоит из введения, обзора литературы, двух глав, отражающих собственные экспериментальные исследования, выводов, списка литературы и приложения. Работа иллюстрирована 000 таблицами, 000 рисунками и 000 схемами. Библиографический список включает 124 отечественных и 64 зарубежных источников.
ОСНОВНОЕ СОДЕРЖАНИЕ РАБОТЫ
1. Характеристика основных объектов и методов исследования
Объектами настоящего исследования являлись образцы древесины лиственницы сибирской и л. даурской; дигидрокверцетин – стандартный образец; биофлавоноидный комплекс диквертин и одноименный препарат «Диквертин»; композиции на базе диквертина, лежащие в основе создаваемых оригинальных комбинированных препаратов. Доминирующий компонент диквертина (90% и более) – дигидрокверцетин. В состав диквертина входят также родственные флавоноиды дигидрокемпферол и нарингенин.
Препарат «Асковертин» является комбинацией диквертина и аскорбиновой кислоты (таблетки, содержащие 0,02 г диквертина в пересчете на 100% содержание дигидрокверцетина и 0,05 г аскорбиновой кислоты). Композиция диквертина и ацетилсалициловой кислоты лежит в основе разрабатываемого препарата «Саливертин» (таблетки покрытые желудочно-резистентной оболочкой, содержащие 0,01 г диквертина в пересчете на 100% содержание дигидрокверцетина и 0,05 г ацетилсалициловой кислоты). Композиция диквертина, аскорбиновой кислоты и b‑каротина является основой БАД «Каровертин» (таблетки, содержащие 0,01 г диквертина в пересчете на 100% содержание дигидрокверцетина, 0,05 г аскорбиновой кислоты и 0,001 г b‑каротина). В исследование включались модельные смеси, субстанции и таблетки одноименных препаратов.
Основным аналитическим методом, используемым в работе, являлся метод ВЭЖХ. Для идентификации и анализа объектов исследования привлекались методы УФ-, ИК-, ЯМР 1Н и ЯМР 13С спектроскопии. Для фракционирования многокомпонентных смесей на стадии пробоподготовки применялась твердофазная экстракция в ее современном техническом исполнении.
2. Методический подход к анализу многокомпонентных смесей на базе диквертина
В состав биофлавоноидного комплекса и вышеуказанных композиций входят разнохарактерные с химической точки зрения соединения: от наиболее полярной и гидрофильной аскорбиновой кислоты, полярных и менее гидрофильных флавоноидов до неполярного и гидрофобного b‑каротина. Эффективное разделение компонентов смеси является основой для их аналитического определения.
Многокомпонентность состава комбинированных препаратов обусловила поиск оптимальных способов анализа и стандартизации их действующих веществ. Методические пути анализа многокомпонентных смесей, использованные в настоящей работе, представлены на схеме 1.
Схема 1. Алгоритм разработки методик анализа смесей на базе диквертина
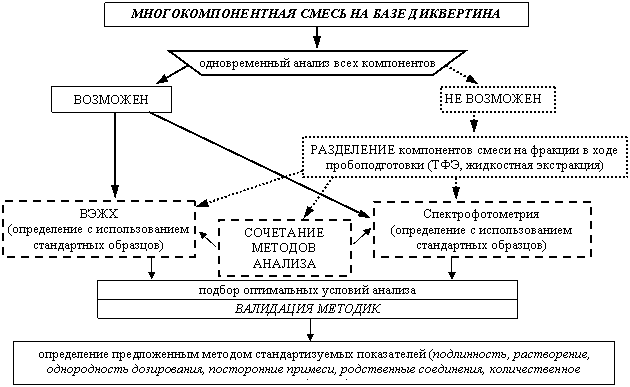

Унификация методик качественного и количественного анализа композиций осуществлялась таким образом, чтобы они служили единой аналитической основой для определения фармакопейных показателей качества препарата: подлинность, посторонние примеси, растворение, однородность дозирования, количественное определение. Большое значение уделено валидации методик, дающей высокую степень уверенности в том, что разработанная методика приводит к результатам, отвечающим установленным критериям приемлемости.
3. Физико-химическая характеристика дигидрокверцетина как стандартного образца
В фармацевтическом анализе для получения точных (правильных) и воспроизводимых результатов анализа ЛС решающее значение имеет применение высокоочищенных и хорошо охарактеризованных стандартных образцов.
Стандартный образец дигидрокверцетина (СО ДГК), получаемый из древесины лиственницы сибирской (Larix sibirica Ledeb.) и л. гмелина, или л. даурской (Larix gmelinii (Rupr.) Rupr., синоним Larix dahurica Turcz.), был разработан в 1996 г. совместно кафедрой органической химии ММА им. и г. Иркутск. За прошедший период проведена работа по совершенствованию способа выделения ДГК из экстракта древесины с использованием ВЭЖХ в полупрепаративном варианте. Экспериментальная работа по получению СО ДГК проведена в , г. Иркутск.
Одна из целей настоящей работы заключалась во всесторонней характеристике физико-химических свойств СО ДГК, доказательстве его строения и степени чистоты.
Индивидуальность и стереохимическое строение ДГК доказывалось с помощью хроматографических (ВЭЖХ) и спектральных (УФ-, ИК-, ЯМР 1Н и ЯМР 13С) методов.
Хроматографический анализ проводили с использованием двух типов сорбентов – октадецилсилановых и цианопропилсилановых – как в изократическом, так и в градиетном режимах элюирования. Подвижной фазой (ПФ) служили смеси ацетонитрила или метанола с водными растворами кислоты (фосфорной, трифтороуксусной или уксусной) с рН 3,0 – 3,5. Создание такой кислотности среды необходимо для подавления диссоциации фенольных гидроксильных групп. Детектирование осуществляли на аналитических длинах волн: при 230 нм для обнаружения соединений с короткой хромофорной системой; при 290 нм – ДГК и флаванонов с близкой структурой и при 370 нм – возможных примесей флавонов. Широкое варьирование условий производили с целью выявления возможных примесей. В качестве оптимальных нами предложены следующие условия ВЭЖХ анализа ДГК: колонка LiChrosper RP-18 5 мкм (250 х 4,6 мм); ПФ: А – ацетонитрил, Б – 2% уксусная кислота. Градиентное элюирование А : Б: 0–4 мин 35 : 65 об.%; 4,1–10 мин 70 : 30 об.%. Скорость потока – 1 мл/мин, аналитическая длина волны 290 нм. В данных условиях эффективность колонки по пику ДГК составляет 5450 т. т.; коэффициент емкости k¢ – 1,6; фактор симметрии пика As – 0,9–1,05; относительное стандартное отклонение площади пика S 0,98%. На всех хроматограммах образцов ДГК содержался единственный пик практически со 100% степенью чистоты.
Степень чистоты ДГК подтверждалась также методом ЯМР. Спектры ЯМР 1Н и ЯМР 13С регистрировали в дейтерированных растворителях – метаноле и диметилсульфоксиде. Отнесение сигналов, их мультиплетность и константы спин-спинового взаимодействия спектра ЯМР 1Н ДГК в ДМСО-D6 в области от 0,5 до 12 м. д. представлены в табл. 1. Суммарная интенсивность всех указанных сигналов соответствует 12 протонам молекулы ДГК С15Н12О7.
Наряду с сигналами протонов, представленных в табл. 1, наблюдаются сигналы растворителя: ДМСО-D6 (квинтет остаточных протонов дейтерированных метильных групп при 2,50 м. д.) и уширенный сигнал воды при 3,34 м. д.; а также незначительные примесные сигналы – уширенный синглет при 1,25 м. д. (0,03 массовых%) и синглет при 2,07 м. д. (0,007 массовых%), который можно отнести к ацетону, используемому в технологическом цикле. В целом, на основании анализа методами ВЭЖХ и ЯМР можно оценить степень чистоты образцов ДГК не ниже 99,96%.
 Также проведено отнесение всех сигналов в спектре ЯМР 13С. Химические сдвиги ядер 13С ДГК в ДМСО-D6 (δ, м. д.): 71,62 (С-3), 83,10 (С-2), 95,02 (С-8), 96, 03 (С-6), 100,53 (С-4а), 115,17 (С-5/), 115,40 (С-2/), 119,41 (С-6/), 128,08 (С-1/), 144,98 (С-3/), 145,81 (С-4/), 162,6 (С-8а), 163,37 (С-5), 166,83 (С-7), 197,78 (С-4).
Также проведено отнесение всех сигналов в спектре ЯМР 13С. Химические сдвиги ядер 13С ДГК в ДМСО-D6 (δ, м. д.): 71,62 (С-3), 83,10 (С-2), 95,02 (С-8), 96, 03 (С-6), 100,53 (С-4а), 115,17 (С-5/), 115,40 (С-2/), 119,41 (С-6/), 128,08 (С-1/), 144,98 (С-3/), 145,81 (С-4/), 162,6 (С-8а), 163,37 (С-5), 166,83 (С-7), 197,78 (С-4).
Таблица 1. Химические сдвиги (d) и константы спин-спинового взаимодействия (J) в ЯМР 1Н спектре СО ДГК в ДМСО-D6 *
(д, д – дублет дублетов, д – дублет, с – синглет, АВ – АВ-система)
Данные спектра | Кольцо С | Кольцо А | Кольцо В | Протоны ОН групп при: | |||||||
Н-2 | Н-3 | Н-6 | Н-8 | Н-2/ | Н-5/ и Н-6/ | С-5 | С-3 | С-7 | С-3/ | С-4/ | |
d, м. д. | 4,973 | 4,483 | 5,898 | 5,848 | 6,868 | 6,72 – 6,76 | 11,87 | 5,712 | 10,78 | 8,98 | 8,93 |
J, Гц | 11,1 | 6,1 и 11,1 | 2,0 | 2,0 | – | 8,2 | – | 6,1 | – | – | – |
Сигнал | д | д, д | д | д | с | АВ | с | д | с | с | с |
* Спектры ЯМР получены и обсуждены старшим научным сотрудником химического факультета МГУ им. , д. хим. н.
Индивидуальность структуры ДГК наиболее полно подтверждена методом ЯМР. Для отнесения сигналов в спектрах ЯМР 1Н и ЯМР 13С наряду с мультиплетностью, интегральной интенсивностью и положением сигналов использовалась информация двумерных экспериментов HSQC и HMBC. Особенно важную информацию дают кросс-пики двумерного эксперимента НМВС, которые позволяют устанавливать корреляцию сигналов 1Н и 13С, находящихся друг от друга через две–три связи. Использование современного подхода позволило впервые разделить и сделать надежное отнесение сигналов протонов всех гидроксильных групп. Ранее не удавалось идентифицировать сигналы протонов гидроксильных групп при С-7, С-3/ и С-4/ сигналы которых перекрывались и наблюдались в виде уширенного пика. В наиболее слабом поле расположен неуширенный сигнал гидроксильной группы 5‑ОН (11,87 м. д.), что свидетельствует об образовании внутримолекулярной водородной связи между 5‑ОН и карбонильной группой. Протон 5‑ОН группы дает отчетливые кросс-пики, соответствующие константам 13С–Н через две, три и четыре связи с атомами углерода С‑5, С‑4а, С‑6, и С‑7. Относительно мало уширен также сигнал протона группы 4¢‑ОН, который имеет кросс‑пики с С‑3¢, С‑4¢, С‑5¢ (рис. 1).
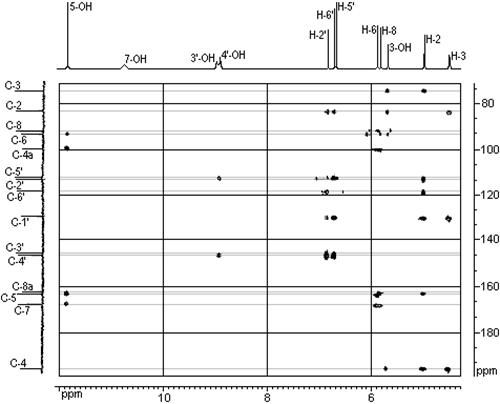
Рис. 1. Двумерный спектр ЯМР дигидрокверцетина: – по оси абсцисс сигналы протонов и их отнесение;– по оси ординат сигналы ядер 13С и их отнесение
Молекулярной специфичностью (за исключением отнесения стероизомеров) обладает и метод ИК-спектроскопии. Для подтверждения индивидуальности исследованного СО ДГК нами предложен ИК‑спектр в виде суспензии в вазелиновом масле (рис. 2).
В высокочастотной области хорошо разрешаются полосы поглощения, обусловленные валентными колебаниями ОН-связей: свободных (3550 см-1); связанных внутримолекулярной (3406 см-1) и межмолекулярной (широкая полоса с максимумом при 3250 см-1) водородными связями.
Валентные колебания связей С–Н в ароматическом кольце проявляются при 3080 см-1, валентные колебания ароматического кольца – при 1615, 1588 см-1. Положение интенсивной полосы валентного колебания карбонильной группы (1644 см-1) свидетельствует об ее участии в водородном связывании. Высокое разрешение полос поглощения в области «отпечатков пальцев» (1300–600 см-1) позволяет использовать ИК-спектр в суспензии в вазелиновом масле для надежной идентификации ДГК (рис. 2).
УФ-спектр СО ДГК в подкисленном этаноле в области длин волн от 230 нм до 380 нм характеризуется наличием полосы поглощения с минимумом при 247±2 нм и максимумом при 290±2 нм, имеющей плечо при 320–327 нм. Концентрации испытуемых растворов (0,001%) отвечала оптическая плотность от 0,590 до 0,640. Для количественной характеристики СО предложен удельный показатель поглощения Е1см1% при длине волны 290,0 нм. Проведено 9 независимых определений удельного показателя трех образцов ДГК. Величина удельного поглощения для этих растворов составила в среднем 623 ± 19.
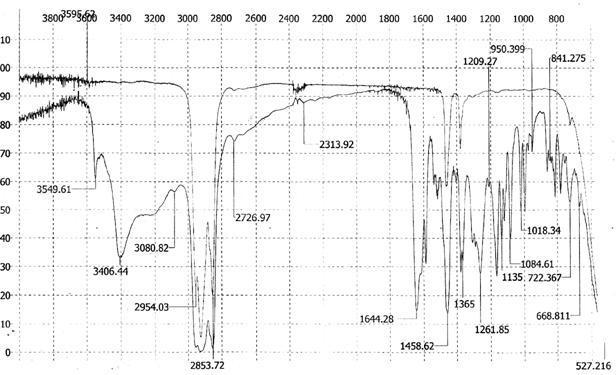
Рис. 2. ИК-спектр дигидрокверцетина в вазелиновом масле
Молекула ДГК содержит два центра хиральности и является оптически активным веществом. Для решения вопроса об относительной конфигурации стереоцентров в молекуле исследуемого дигидрокверцетина ключевую роль играет величина константы спин-спинового взаимодействия (J) протонов Н-2 и Н-3. По данным ЯМР 1Н спектра эта константа равна 11,1 Гц, что свидетельствует об их транс-расположении. Такую относительную конфигурацию имеют два стереоизомера дигидрокверцетина – 2R,3R-изомер и его энантиомер – 2S,3S-изомер. Учитывая сумму установленных в настоящее время физико-химических характеристик для всех четырех стереоизомерных форм ДГК, можно воспользоваться сравнением со специфическим сочетанием для каждого стереоизомера двух характеристик: величины константы J и направления угла оптического вращения.
Исследуемый в нашей работе ДГК имеет положительное значение удельного вращения, что в сочетании со значением константы J 11,1 Гц позволяет считать его изомером с 2R,3R-конфигурацией.
С помощью комплекса физико-химических методов установлено, что СО ДГК представляет собой индивидуальное соединение высокой степени чистоты, отвечающее структуре (2R,3R)-2,3-дигидро-3,5,7-тригидрокси-2-(3,4-дигидроксифенил)-4Н-1-бензопиран-4-она.
На базе высокоразрешающей аппаратуры усовершенствованы спектральные и хроматографические методики анализа, обеспечивающие достоверность проверки подлинности и чистоты ДГК, которые легли в основу соответствующей ФС для регистрации ДГК в качестве государственного стандартного образца.
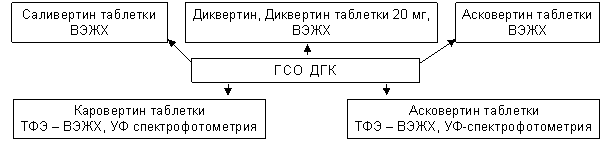
ГСО ДГК использовался в нашей работе для количественного определения модельных смесей, субстанций и лекарственных форм исследуемых объектов (схема 2).
Схема 2. Использование ГСО ДГК для количественного определения препаратов на базе диквертина

4. Анализ и стандартизация диквертина в субстанции, таблетках и растительном сырье
В состав субстанции диквертина (ДКВ) входят ДГК (90% и более), родственные соединения флаванонового ряда – дигидрокемпферол и нарингенин, допускается наличие флавонола кверцетина. Суммарное содержание дигидрокемпферола, нарингенина и кверцетина в субстанции должно быть не более 10%. В древесине листвинницы в незначительных количествах присутствуют пиностробин, пиноцембрин и пинобаксин.
Учитывая многокомпонентный состав ДКВ, оптимальным для его анализа является метод ВЭЖХ. Общим в структуре флавоноидных компонентов ДКВ является наличие малополярной ароматической углеродной и гетероуглеродной основы, несущей более полярные гидроксильные заместители. Такой тип структуры предопределяет использование обращенно-фазного варианта ВЭЖХ.
Пробоподготовка субстанции и таблеток ДКВ заключалась в непосредственном растворении образцов в ацетонитриле, фильтрации и дальнейшем разбавлении смесью растворителей, используемой в качестве ПФ. Нерастворимые в ацетонитриле вспомогательные вещества удалялись фильтрованием. Установлено, что вспомогательные вещества, растворимые в ацетонитриле, не мешают определению компонентов ДКВ.
При выборе аналитической длины волны изучены УФ-спектры ДКВ в ацетонитриле с добавлением водного раствора кислоты или щелочи в интервале pH от 2 до 8. В интервале pH от 2 до 4 спектр ДКВ, как и спектр ДГК, характеризуется максимумом полосы поглощения при 290±2 нм, имеющий плечо при 320–327 нм. При увеличении pH от 4 до 8 наблюдается уменьшение интенсивности поглощения при 290 нм и увеличение при 325 нм, что обусловлено ионизацией фенольных гидроксильных групп. Наилучшая воспроизводимость спектров достигается в интервале pH 2–3, когда ДГК находится в неионизированном состоянии. Это обстоятельство учитывалось при выборе состава ПФ. Дигидрокемпферол и нарингенин имеют сходную с ДГК хромофорную систему и, соответственно, подобный УФ-спектр. Кверцетин (флавонол) имеет максимум поглощения при 370 нм. Современные детекторы позволяют проводить обсчет результатов сразу для нескольких длин волн. В то же время чувствительность УФ детекторов такова, что, даже используя в качестве аналитической только длину волны 290 нм, можно обнаружить кверцетин (если он имеется) в образце ДКВ вплоть до его содержания 0,01% по массе.
Выбор обращенно-фазного сорбента осуществлен на основе анализа эффективности разделения компонентов ДКВ на этил-, октил - и октадецилсилановых сорбентах с использованием модельных смесей, содержащих ванилин в качестве внутреннего стандарта. В составе ПФ использовали метанол или ацетонитрил и 2% уксусную кислоту. Расчет хроматографических параметров показывает преимущество октадецилсиланового сорбента по эффективности, симметрии пика и воспроизводимости площади пика (табл. 2).
Таблица 2. Сравнение некоторых хроматографических параметров анализа ДКВ (при 5 повторных определениях)
Определяемый показатель | Колонки | ||
LiChrosorb 100 RP-2 250 мм х 4,6 мм; 5 мкм | LiChrosorb 100 RP-8 250 мм х 4,6 мм; 5 мкм | LiChrosorb 100 RP мм х 4,6 мм; 5 мкм | |
N по пику ДГК, т. т. | 815, 860, 900, 840, 810 | 3900, 3850, 3800, 3600, 3700 | 4300, 4400, 4500,4800,4900 |
RS пиков ДГК и ванилина | 2,1; 2,0; 2,1; 2,0; 2,1 | 2,0;2,2; 2,0; 2,1; 2,1 | 2,7; 2,6; 2,5; 2,5; 2,5 |
S площади пика ДГК, % | 1,8 ± 0,2 | 1,5 ± 0,2 | 1,1 ± 0,1 |
AS пика ДГК | 0,9; 1,5; 2,0; 0,7; 1,2 | 0,8; 1,2; 1,5; 1,2; 1,1 | 1,0; 1,1; 0,8; 0,9; 1,0 |
Изократический режим элюирования не позволяет оптимизировать хроматографический процесс для каждого компонента ДКВ. В ПФ ацетонитрил – 2% уксусная кислота (30 : 70 об.%) оптимизированы условия для ДГК и дигидрокемпферола, но на хроматограммах наблюдается асимметрия пиков нарингенина и кверцетина, имеющих большую величину коэффициетна емкости k¢. При резком увеличении органического модификатора ПФ (ацетонитрил – 2% уксусная кислота 70 : 30 об.%), ДГК элюируется практически с мертвым временем колонки, но для пиков нарингенина и кверцетина это соотношение оптимально. Поэтому, для оптимизации хроматографического процесса подобран градиентный режим элюирования: колонка 250 мм х 4,6 мм; сорбент LiChrospher 100, RP-18, 5 мкм; аналитическая длина волны 290 нм; ПФ– ацетонитрил (А), кислота уксусная 2,0% (Б); условия градиента (время/отношение А к Б (об.%)): 0 – 4,0 мин / 35 : 65, 4,1 – 10,0 мин / 70 : 30; скорость потока ПФ 1 мл/мин; температура колонки 20 °С
Предлагаемые условия оптимизированы по хроматографическим параметрам (табл. 3). Значения коэффициента емкости k¢ находятся в диапазоне от 0,532 для ДГК до 2,902 для кверцетина. Оптимальными являются значения параметров селективности a и разрешающей способности RS, служащие мерой качества разделения.
Эффективность хроматографической колонки (N), рассчитанная для пика ДГК, составила от 4325 до 5100 теоретических тарелок, коэффициент ассиметрии пика (AS) от 0,94 до 1,05. Относительное стандартное отклонение площади пика ДГК (S) составило 1,0%. Хроматографические параметры свидетельствуют о пригодности хроматографической системы. Методика валидирована по соответствующим параметрам, подтверждающим ее специфичность, точность и воспроизводимость, линейную зависимость в аналитической области, что позволяет использовать ее для достоверной оценки качества субстанции и препарата диквертин.
Таблица 3. Параметры хроматографического разделения компонентов диквертина (среднее из 5 определений)
Соединение | Эффективность N, т. т. | Коэффициент емкости, k¢ | Селективность, a | Разрешающая способность, RS |
Дигидрокверцетин | 5050 | 0,532 | ||
1,859 | 1,86 | |||
Дигидрокемферол | 3210 | 0,989 | ||
2,612 | 15,4 | |||
Нарингенин | 4420 | 2,585 | ||
1,124 | 1,89 | |||
Кверцетин | 5320 | 2,902 |
При разработке методики анализа большое внимание уделялось унификации условий анализа с целью возможности сквозной стандартизации диквертина от растительного сырья до лекарственной формы по фармакопейным тестам (схема 3).
Схема 3. Унификация ВЭЖХ методики анализа диквертина
Анализ ДКВ методом ВЭЖХ | ||||
| ||||
Стандартизуемые показатели | ||||
|
|
|
|
|
ПОДЛИННОСТЬ | ПОСТОРОННИЕ ПРИМЕСИ | РОДСТВЕННЫЕ СОЕДИНЕНИЯ | ОДНОРОДНОСТЬ ДОЗИРОВАНИЯ | КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ |
растительное сырье субстанция таблетки | субстанция таблетки | субстанция таблетки | субстанция таблетки | растительное сырье субстанция таблетки |
Подтверждение подлинности ДКВ производится путем сопоставления хроматограмм образцов субстанции или таблеток с хроматограммой СО ДГК. Наряду с пиком ДГК в образцах ДКВ содержатся пики, соответствующие дигидрокемпферолу и нарингенину с временами удерживания относительно СО ДГК 1,3 ± 0,1 и 2,4 ± 0,1 соответственно. Возможно наличие пика, соответствующего кверцетину с относительным временем удерживания 2,7 ± 0,1.
Содержание родственных соединений и ДГК в образцах ДКВ в процентах рассчитывается на основании площадей соответствующих пиков и пика ГСО ДГК. Суммарное содержание дигидрокемпферола, нарингенина и кверцетина в образце должно быть не более 10,0%, содержание ДГК – не менее 90,0%.
Стандартизация растительного сырья, содержащего ДКВ. В целях сквозной стандартизации ДКВ от растительного сырья до препарата разработанная методика адаптирована для анализа содержания ДГК в цельном и измельченном ЛРС – древесине лиственницы.
С этой целью разработана специальная процедура пробоподготовки, направленная на освобождение исследуемой пробы от ряда примесей, способных не только помешать анализу, но и опасных с позиции сохранности сорбента колонки и хроматографического оборудования. В методику пробоподготовки к анализу была введена стадия твердофазной экстракции (ТФЭ). Выявлено, что оптимальным сорбентом для проведения ТФЭ явились патроны Nexus 60 mg (Varian). Путем последовательного промывания патрона с нанесенной пробой растворителями различной полярности производилось удаление примесей – неполярных (элюированием хлороформом и бензолом) и полярных (элюированием 2% водным раствором уксусной кислоты). Подбор экспериментальных условий элюирования примесей без десорбции ДГК проводили под контролем чувствительной цианидиновой пробы и с помощью ВЭЖХ.
Для элюирования ДГК с патрона достаточно 1 мл ацетонитрила. Анализ содержания ДГК в древесине лиственницы проводили по вышеописанной методике.
Установлено, что содержание ДГК в древесине лиственницы варьирует в широком интервале от 0,5% до 3,0 – 3,5% (редко до 4,0 – 4,5%). Это связано с разными географическими зонами произрастания. Важно отметить, что по количественному содержанию ДГК в древесине нет принципиального различия между двумя ботаническими видами лиственницы – сибирской и даурской.
5. Анализ и стандартизация комбинированных препаратов на базе диквертина
5.1. Способы анализа многокомпонентных препаратов на основе диквертина с использованием метода твердофазной экстракции
Для стандартизации асковертина, саливертина и каровертина необходимо было решить задачи пробоподготовки и анализа действующих веществ - ДГК и аскорбиновой кислоты (АК), ДГК и ацетилсалициловой кислоты (АСК), ДГК и АК и β-каротина соответственно.
Непосредственное одновременное спектрофотометрическое определение всех компонентов комбинированных препаратов в одной пробе затруднено тем, что получаемая при их совместном присутствии кривая УФ-спектра представляет собой спектр наложения, в котором невозможно выделить области индивидуального поглощения каждого из компонентов. Кроме того, спектры действующих веществ в неионизированном и ионизированном состоянии значительно различаются и необходимо было выбрать интервал рН, в котором воспроизводимость спектра наибольшая. Так, анализ водных и водно-метанольных растворов АК методом УФ-спектроскопии показал, что в течение короткого промежутка времени происходит изменение интенсивности оптического поглощения. В водных растворах кислот – 0,1% H3PO4 или 0,03% CF3COOH – с pH 2,5–2,7 (степень ионизации, вычисленная по данным pKa АК = 4,04 и pH составляет не более 2%), а также при добавлении к растворам кислот до 30% метанола по объему оптическая плотность АК не изменяется в течение 2 ч, что достаточно для анализа. Стабильность действующих веществ в той или иной среде учитывалась при выборе экстрагентов и элюентов.
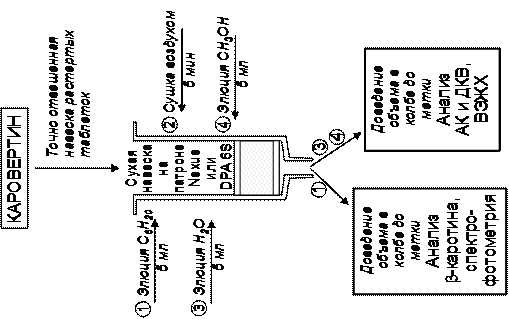
Для каждого из перечисленных препаратов предложены варианты анализа, основанные на предварительном разделении компонентов методом ТФЭ (рис. 3). Анализируемые смеси пропускали через колонку, заполненную соответствующим адсорбентом, на которой сорбировались отдельные компоненты. В наибольшей степени данной цели отвечали универсальные патроны для ТФЭ Nexus (Varian) и патроны с полиамидным сорбентом DPA-6S (Supelco). Разделение компонентов достигалось элюированием подходящим растворителем.
|
Достоинством способа анализа, основанного на разделении компонентов методом ТФЭ и их спектрофотометрическом определении, является экспресность и применение доступного, недорогого оборудования. Вместе с тем, очевидны и недостатки метода. Не учитывается вклад в поглощение ДГК родственных соединений – дигидрокемпферола и нарингенина, имеющих аналогичные спектральные характеристики. Таким образом, можно проводить анализ только совокупности флавоноидных компонентов ДКВ, а не ДГК. При анализе АСК не учитывается и не анализируется примесь СК, а ее содержание в препаратах АСК регламентируется. В случае асковертина оценка эффективности ТФЭ показала, что не происходит полного разделения АК и флавоноидов, до 3–4% флавоноидов от их общего содержания в таблетке не сорбируется на патроне ТФЭ и находится в водном растворе. Поэтому данный подход не является оптимальным для стандартизации асковертина и саливертина и для этих препаратов безусловный интерес представляла разработка универсальной методики одновременного определения действующих веществ, родственных соединений и примесей с использованием метода ВЭЖХ.
В то же время для стандартизации каровертина данный вариант анализа является оптимальным, так как в альтернативном случае пришлось бы разрабатывать два способа ВЭЖХ анализа – один для АК и флавоноидов и другой для β-каротина, учитывая, что их анализ требует различного сочетания подвижных и неподвижных фаз. Использование ТФЭ (рис. 3в) позволяет отделить АК и проанализировать ее спектрофотометрически, выделить и проанализировать методом ВЭЖХ компоненты ДКВ. β-Каротин экстрагируется из исследуемого образца гексаном и анализируется спектрофотометрически при длине волны 465±4 нм с использованием удельного показателя поглощения равного 2335. Предлагаемый вариант анализа с использованием ТФЭ характеризуется высокой селективностью, чувствительностью и воспроизводимостью.
![]()
![]()
![]()
![]()
![]()

![]()
![]()


5.2. Разработка унифицированной методики одновременного определения компонентов препарата Асковертин методом ВЭЖХ
В результате исследования различных вариантов пробоподготовки к анализу методом ВЭЖХ выбран простой, эффективный способ жидкостной экстракции. Компоненты ДКВ полностью извлекаются метанолом, при последующем добавлении к пробе 0,03% раствора ТФУ происходит полное растворение АК. Определяемые компоненты АК и ДГК устойчивы в растворах ТФУ и ПФ в течение 2 ч и более.
Поиск лучшего варианта хроматографических условий показал, что оптимальное разделение высокополярной АК и менее полярных и гидрофильных флавоноидов ДКВ достигается на сорбентах с привитой циано–фазой в режиме обращенно-фазного элюирования (сорбент Zorbax CN, 5 мкм; ПФ метанол – 0,03% трифтороуксусная кислота 21 : 79 об.%). При данной кислотности ПФ (pH среды 2,7) все компоненты асковертина находятся в неионизированной форме и наряду с неспецифическими взаимодействиями «сорбат-сорбент» появляются специфические – водородные связи между сорбатом и нитрильной группой сорбента. Таким образом, селективность данной хроматографической системы отличается от селективности традиционных обращенно-фазных алкильных сорбентов, при разгонке на которых АК элюируется практически с мертвым объемом.
Методика одновременного анализа АК и флавоноидов ДКВ оптимизирована по хроматографическим параметрам. Пики родственных ДГК соединений, входящих в состав субстанции ДКВ, четко различаются от пиков действующих веществ и имеют следующие показатели времени удерживания относительно СО ДГК: дигидрокемпферол 1,3 ± 0,1 и нарингенин 1,9 ± 0,1. Эффективность хроматографической колонки (N) варьирует по пику АК от 1554 до 1577 и по пику ДГК от 1354 до 1370 т. т. Разрешающая способность R для пары АК–ДГК составляет 1,3, для пары ДГК–дигидрокемпферол 1,1, для пары дигирокемпферол–нарингенин 2,5. Коэффициент асимметрии пика варьирует от 0,8 до 0,9 для пика АК и от 0,85 до 1,08 для пика ДГК. Относительное стандартное отклонение площади пика (S) составило 2,49% ± (n = 5) для площади пика АК и 1,32% ± (n = 5) для площади пика ДГК. Все приведённые параметры характеризуют пригодность хроматографической системы для анализа асковертина. Для проведения количественного анализа компонентов асковертина АК и ДГК методика валидирована по линейности, воспроизводимости, правильности.
Унификация методики ВЭЖХ анализа осуществлена таким образом, чтобы она служила единой аналитической базой для испытаний на подлинность, родственные соединения, растворение, однородность дозирования и для количественного определения АК и ДГК в таблетках. Подлинность: на хроматограмме полученной методом ВЭЖХ в предложенных условиях, должны содержаться пики ДГК и АК, совпадающие по времени удерживания с пиками ГСО ДГК и РСО АК, а также должны обнаруживаться родственные соединения – дигидрокемпферол и нарингенин.
На рисунке 4 приведены результаты теста «растворение», который введен только для ДГК как наименее растворимого из двух действующих веществ. Определение проводили в соответствии с требованиями ОФС 42‑0003‑00. По данным анализа 5 серий препарата через 20 мин количество ДГК в растворе достигало максимума и составляло не менее 80% от его номинального содержания в таблетке.
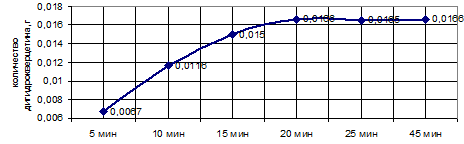
Рис. 4. Профиль высвобождения ДГК из таблеток асковертина
По результатам теста «однородность дозирования» отклонение содержания ДГК в одной таблетке составило от + 7 до – 6,5% от среднего содержания; отклонение содержания АК одной таблетке составило от + 7,2 до – 6,0% от среднего содержания, что удовлетворяет требованиям ГФ XI.
Результаты количественного определения АК и ДГК 5 опытно-промышленных партий таблеток «Асковертин», выработанных , приведены в табл. 4.
Таблица 4. Результаты анализов образцов таблеток Асковертина, изготовленных
Серия | Количественное определение в таблетке (ВЭЖХ): | |
дигидрокверцетина, от 0,0185 г до 0,0215 г | аскорбиновой кислоты от 0,0462 г до 0,0537 г | |
010503 | 0,0194 | 0,0485 |
020603 | 0,0196 | 0,0510 |
030703 | 0,0200 | 0,0492 |
040803 | 0,0201 | 0,0515 |
050903 | 0,0187 | 0,0490 |
Таким образом, разработанная методика анализа действующих веществ асковертина методом ВЭЖХ является универсальной и может быть использована для определения фармакопейных показателей, лежащих в основе оценки качества препарата.
5.3. Разработка унифицированной методики анализа компонентов саливертина методом ВЭЖХ
Основная цель в создании такой методики заключалась в разработке оптимальных условий одновременного определения действующих веществ саливертина – ДГК и АСК, родственных соединений биофлавоноидного комплекса ДКВ и посторонней примеси СК.
Хорошее разделение всех компонентов композиции ДКВ и АСК удалось реализовать в обращенно-фазном варианте в ПФ метанол-водный раствор кислоты в условиях градиентного элюирования. В качестве аналитической выбрана длина волны 280 нм, при которой АСК, СК и компоненты ДКВ имеют интенсивное поглощение.
Разработка состава ПФ включала выбор органического компонента и водного раствора кислоты. Для данной многокомпонентной смеси преимущество, по сравнению с ацетонитрилом, имеет метанол, как более «мягкий» элюент, имеющий более низкую элюирующую способность. Для выбора лучших параметров удерживания изучено влияние величины ПФ и типа кислоты на эффективность, разрешающую способность и размывание хроматографической зоны.
В таблице 5 представлены параметры пиков ДГК и АСК, полученные при использовании ПФ метанол-водный раствор кислоты в изократическом режиме 45 : 55 об.%. Оптимальное значение коэффициента ёмкости k¢ реализуется только для ПФ с 1% уксусной кислотой или 0,01% фосфорной кислотой. Наиболее высокая симметрия пиков ДГК и АСК достигается при использовании ПФ с ортофосфорной кислотой или трифтороуксусной кислотой (коэффициент ассиметрии пиков ДГК и АСК составляет в ПФ с H3PO4 0,93–0,94 и 0,91–1,05, соответственно, тогда как при использовании 1% CH3COOH 0,89 и 0,84). Критерий разделения пиков RS ДГК/АСК и селективность aДГК/АСК при использовании ортофосфорной кислоты находится в оптимальном диапазоне значений, что предопределило включение данной кислоты в состав ПФ.
Таблица 5. Выбор оптимального кислотного модификатора ПФ
Состав ПФ | CH3OH : 5% CH3COOH pH 3,51 | CH3OH : 2% CH3COOH pH 3,68 | CH3OH : 1% CH3COOH pH 3,68 | CH3OH : 0,01% H3PO4 pH 3,25 | CH3OH : 0,03% CF3COOH pH 2,88 |
k¢ДГК | 0,682 | 0,902 | 0,964 | 1,020 | 0,890 |
k¢АСК | 1,07 | 1,27 | 1,27 | 1,32 | 1,21 |
aДГК/АСК | 1,57 | 1,39 | 1,30 | 1,29 | 1,39 |
NДГК | 5202 | 4842 | 5305 | 5202 | 4389 |
NАСК | 7698 | 7025 | 7844 | 7516 | 6505 |
RS ДГК/АСК | 3,73 | 3,10 | 2,71 | 2,9 | 2,9 |
ASДГК | 0,77 | 1,13 | 0,89 | 0,93 | 0,91 |
ASАСК | 0,94 | 0,95 | 0,84 | 0,94 | 1,05 |
Для оптимизации хроматографического процесса по каждому компоненту саливертина предпочтительным является градиентный режим разгонки: фаза А – CH3OH, фаза Б – 0,01% H3PO4; в начальный момент соотношение А : Б – 45 : 55 об.%, к седьмой минуте разгонки 48 : 52 об.%, к 15 минуте 80 : 20 об.%. Хроматограмма образца таблетки саливертина представлена на рис. 5, а параметры разделения компонентов в табл. 6. Как видно из представленных данных все параметры находятся в оптимальном диапазоне.
Лекарственной формой саливертина являются таблетки покрытые кишечно-растворимой оболочкой. Наряду со вспомогательными веществами ядра (мелкокристаллическая целлюлоза, лактоза, картофельный крахмал и стеарат кальция) они содержат вспомогательные вещества оболочки (колликут МАЕ-100Р, полиэтиленоксид 400, диоксид титана). Процесс пробоподготовки оптимизирован таким образом, чтобы вспомогательные вещества ядра и оболочки не извлекались в анализируемый раствор. Поиск подходящего экстрагента привел к выбору в качестве такового ацетонитрила. Установлено, что в нем плохо растворимы компоненты оболочки таблетки, которые удаляют фильтрованием, но хорошо растворимы действующие вещества. Полученный раствор разбавляют ПФ А до удобного для анализа уровня концентраций действующих веществ.
|
|
|
|
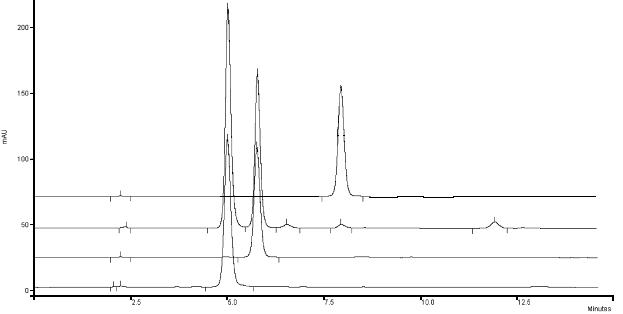
Рис. 5. Хроматограммы, иллюстрирующие селективность определения дигидрокверцетина, ацетилсалициловой кислоты и салициловой кислоты в таблетках саливертина
а – ГСО ДГК; б – РСО АСК; в – таблетки саливертина, порядок выхода веществ: ДГК (5,3); АСК (6,1); дигидрокемпферол (7,3); СК (8,5); нарингенин (12,8); г – РСО СК.
Таблица 6. Хроматографические параметры разделения компонентов смеси АСК и ДКВ (среднее из n=9)
Соединение | N | k¢ | a | RS |
ДГК | 5000 | 1,020 | ||
1,29 | 2,9 | |||
АСК | 7000 | 1,32 | ||
1,49 | 2,52 | |||
Дигидрокемпферол | 4800 | 2,12 | ||
1,11 | 1,32 | |||
Салициловая кислота | 7900 | 2,53 | ||
1,82 | 11,24 | |||
Нарингенин | 6510 | 4,27 |
Разработанная методика позволяет проводить качественный и количественный анализ саливертина с использованием внешних стандартов образцов – ГСО ДГК, РСО АСК и СК. Относительное стандартное отклонение площади пика ДГК составляет 2,37%, АСК 1,22% .Универсальность методики ВЭЖХ анализа саливерина позволяет использовать её в фармакопейных тестах «подлинность», «количественное определение», «растворение», «однородность дозирования», «посторонние примеси».
Поскольку саливертин относится ко 2-й группе ГЛФ – желудочно-резистентные таблетки, то в соответствии с фармакопейными требованиями тест «растворение» проводится в две стадии – в кислой, а затем нейтральной среде. Тест «растворение» может быть введен только для ДГК, как наименее растворимого из двух действующих веществ. Количество перешедшего в раствор ДГК определяли методом ВЭЖХ по представленной методике, в сравнении с раствором ГСО ДГК в подвижной фазе. Таким образом, для тестов «растворение» и «количественное определение» предложена единая методика ВЭЖХ, а в качестве стандарта один и тот же раствор ГСО ДГК. По данным анализа 5 серий препарата на 1-й стадии (кислотной) в раствор через 60 мин переходило не более 10% ДГК, на 2-й стадии (буферной) в растворе через 45 мин определялось не менее 70% ДГК, что соответствует требованиям ГФ XI.
Количественный анализ ДГК и АСК с использованием стандартных образцов по данной единой методике ВЭЖХ лежит и в основе теста «однородность дозирования».
В препаратах, содержащих АСК в качестве посторонней примеси может присутствовать СК. Она образуется в результате гидролиза АСК на технологических стадиях производства и при нарушении условий хранения. В различных фармакопеях допустимые уровни нормирования примеси СК в лекарственных формах АСК несколько различаются. По Европейской Фармакопее содержание СК в таблетках АСК должно быть не более 0,15%, а по Фармакопее США – не более 0,3%. В таблетках аспирина покрытых оболочкой в Фармакопее США нормируется допустимая примесь СК не более 3%.
Обнаружение примеси СК в препарате осуществляется на основании совпадения на хроматограммах времен удерживания пика в образцах с таковым для РСО СК. Методика позволяет анализировать СК в таблетках без оболочки и в таблетках покрытых оболочкой. Для таблеток покрытых оболочкой анализ СК рекомендуется проводить, используя разбавленный раствор, одновременно с анализом действующих компонентов препарата, а не покрытых желудочно-резистентной оболочкой более концентрированный раствор.
По результатам анализа таблеток 5 серий препарата саливертина установлено, что содержание СК изменялось в интервале 0,0009 – 0,0014 г. Ни в одной из анализируемых серий содержание примеси СК не превышало допустимое значение (не более 3%).
Разработанная методика оценки качества саливертина с использованием ВЭЖХ испытана в анализе модельных смесей и таблеток двух лабораторных партий. Методика нашла отражение в подготавливаемой фармакопейной документации для регистрации препарата саливертина «подлинность», «однородность дозирования», «растворение», «посторонние примеси», «количественное определение».
6. Валидация методик анализа диквертина, асковертина и саливертина
Важнейшим критерием оценки аналитической методики служит доказательство её валидности, включающей взаимосвязанную систему характеристик – специфичность, пригодность хроматографической системы, линейность, правильность и воспроизводимость.
С этой целью выполнено экспериментальное исследование по изучению комплекса валидационных характеристик разработанных методик определения действующих веществ указанных препаратов методом ВЭЖХ. Пригодность хроматографической системы для анализа каждого препарата подтверждена оптимальными значениями хроматографических параметров, представленными выше.
Специфичность методики основана на возможности достоверно определять количественное содержание действующих веществ в таблетке в присутствии вспомогательных веществ и родственных соединений. Процесс пробоподготовки оптимизирован таким образом, что вспомогательные вещества ядра и оболочки не извлекаются в анализируемый раствор. На хроматограммах модельной смеси, содержащей вспомогательные вещества без действующих веществ (плацебо), отсутствуют пики, мешающие определению действующих веществ. Тождественность действующих веществ препаратов ДГК, АК, АСК и СК подтверждалась совпадением времен удерживания анализируемых компонентов таблетки и стандартных образцов. Пики родственных соединений, входящих в состав субстанции диквертин, хорошо разделяются с пиками действующих веществ.
Линейная зависимость методики отражает пропорциональность возрастания площади пика на хроматограмме при возрастании количества анализируемых веществ в испытуемых образцах. Данная валидационная характеристика исследовалась на модельных смесях в интервале 70–130% от заявленного содержания действующих компонентов в таблетках. Зависимость аналитического сигнала (условных единиц площади пика) от содержания анализируемых веществ (в г) описаны уравнением регрессии y = bx + a (табл. 7). Согласно полученным результатам все линейные зависимости характеризуются высоким коэффициентом корреляции в интервале 70–130% декларируемой величины. Этот интервал можно определить как аналитическую область методики.
Правильность (точность) методики показывает систематические погрешности метода и выражается как процент регенерации точно взвешенного количества анализируемого образца. Правильность разработанных методик установлена по результатам анализа модельных смесей с использованием СО для трех повторностей определений семи аналитических концентраций. В таблице 8 продемонстрированы результаты данной характеристики валидности на примере саливертина. Как видно из представленных данных, методика обладает удовлетворительной точностью. Полученные аналогичным образом результаты для препарата асковертина показали, что средний процент регенерации составляет для АК 99,1%, для ДГК – 99,2%. Результаты оценки правильности, полученные для всех методик, находятся в интервале 97–103%.
Таблица 7. Параметры линейной регрессии зависимостей площадей пиков от содержания анализируемых веществ в модельных смесях
Действующее вещество | Отрезок а | Наклон b | Коэффициент корреляции |
ДИКВЕРТИН | |||
ДГК | 127000 | 8,647 | 0,9993 |
АСКОВЕРТИН | |||
ДГК | 7257,3571 | 7,0212 | 0.9984 |
АК | 6554,3571 | 3,8295 | 0,9998 |
САЛИВЕРТИН | |||
ДГК | 11310 | 6,16 | 0,9989 |
АСК | 340660 | 8,88 | 0,9993 |
СК | 1050 | 9,63 | 0,9886 |
Таблица 8. Оценка правильности методики определения действующих компонентов саливертина
Действующих веществ от заявленного в таблетке, % | Состав в модельной смеси, г | Найдено*,г | Регенерация*,% | |||
ДГК | АСК | ДГК | АСК | ДГК | АСК | |
70 | 0,0072 | 0,0358 | 0,0071 | 0,0357 | 98,6 | 99,7 |
80 | 0,0080 | 0,0407 | 0,0082 | 0,0405 | 102,5 | 99,5 |
90 | 0,0091 | 0,0455 | 0,0090 | 0,0454 | 98,9 | 99,7 |
100 | 0,0101 | 0,0506 | 0,0100 | 0,0507 | 99,0 | 100,2 |
110 | 0,0112 | 0,0555 | 0,0112 | 0,0558 | 101,8 | 100,5 |
120 | 0,0121 | 0,0602 | 0,0121 | 0,0600 | 100,0 | 99,7 |
130 | 0,0134 | 0,0653 | 0,0133 | 0,0652 | 99,3 | 99,8 |
* - среднее из 3 определений
Воспроизводимость аналитической методики характеризовалась степенью совпадения результатов индивидуальных определений при многократном использовании. По представленным в таблице 9 параметрам (величины стандартного отклонения, доверительный интервал) можно сделать заключение о хорошей воспроизводимости методик анализа асковертина и саливертина. Относительная ошибка среднего результата определения ДГК составляет 2,73 и 1,97% соответственно, АК 1,70%, АСК – 1,74% и СК 2,9%.
Таблица 9. Оценка воспроизводимости методик анализа асковертина и саливертина
хi, мг | n | хср±Dхср | S | eср | Р, f | хi, мг | n | хср±Dхср | S | eср |
АСКОВЕРТИН | ||||||||||
по ДГК | по АК | |||||||||
18,90; 19,27; 19,54; 19,58; 19,84; 20,20; 20,45; 20,85; 21,04 | 9 | 19,96±0,5452 | 0,724 | 2,73 | 2,26 | 48,50; 48,85; 48,93; 49,00; 49,20; 50,10; 50,75; 51,23; 51,47 | 9 | 49,78±0,85 | 1,3 | 1,70 |
САЛИВЕРТИН | ||||||||||
по ДГК | по АСК | |||||||||
10,1; 10,2; 9,8; 9,7; 10,3; 9,5; 9,9; 9,8; 10,1 | 9 | 9,93±0,1959 | 0,260 | 1,97 | 2,26 | 48,51; 48,96; 51,33; 48,80; 50,10; 50,85; 49,01; 49,23; 51,47 | 9 | 49,8±0,87 | 1,15 | 1,74 |
По валидационным характеристикам разработанные методики являются специфичными для определения содержания действующих веществ, характеризуются корректной точностью и воспроизводимостью, линейной зависимостью в аналитической области ± 30% по отношению к заявленному содержанию действующих веществ в таблетках, что позволяет использовать их для достоверной оценки качества препаратов.
ВЫВОДЫ
1. Разработан единый аналитический подход к стандартизации и оценке качества в цепи объектов: древесина лиственницы – биофлавоноидный комплекс диквертин – комбинированные препараты на базе диквертина: асковертин (с аскорбиновой кислотой), саливертин (с ацетилсалициловой кислотой), БАД каровертин (с аскорбиновой кислотой и b-каротином). Аналитический подход, включающий этапы соответствующей пробоподготовки и анализа методом ВЭЖХ, применим для определения фармакопейных показателей как субстанций, так и лекарственных форм.
2. С помощью комплекса физико-химических методов установлено, что стандартный образец дигидрокверцетина (производства , г. Иркутск) представляет собой индивидуальное соединение высокой степени чистоты, отвечающее структуре (2R,3R)-2,3-дигидро-3,5,7-тригидрокси-2-(3,4-дигидроксифенил)-4Н-1-бензопиран-4-она. Предложены спектральные и хроматографические методики анализа, обеспечивающие достоверность подлинности (ИК-, ЯМР 1Н и ЯМР 13С спектры) и чистоты (ВЭЖХ, ЯМР 1Н и УФ-спектроскопия) дигидрокверцетина. Исследованные образцы дигидрокверцетина соответствуют фармакопейным требованиям, предъявляемым к государственным стандартным образцам, и могут быть использованы для анализа лекарственных средств и лекарственного растительного сырья.
3. Определены оптимальные условия разделения и анализа флавоноидных компонентов диквертина с использованием обращенно-фазного варианта ВЭЖХ на октадецилсилановых сорбентах в градиентном режиме элюирования. Предложена усовершенствованная методика стандартизации субстанции и таблеток диквертина с применением стандартного образца дигидрокверцетина и обосновано ее применение для анализа основного компонента дигидрокверцетина и родственных соединений – дигидрокемпферола и нарингенина.
4. Установлено, что оптимальное разделение полярной и гидрофильной аскорбиновой кислоты и менее полярных и более гидрофобных флавоноидных компонентов диквертина достигается на сорбентах с привитой циано-фазой в режиме обращенно-фазного элюирования. На основе изученных хроматографических характеристик композиции диквертина и аскорбиновой кислоты предложена унифицированная методика стандартизации субстанции и таблеток препарата асковертина.
5. Разработаны эффективные способы пробоподготовки и одновременного анализа методом ВЭЖХ ацетилсалициловой кислоты, флавоноидных компонентов диквертина и салициловой кислоты (как возможной примеси) в таблетках саливертина, покрытых желудочно-резистентной оболочкой. В предлагаемых хроматографических условиях эффективность колонки по пику дигидрокверцетина составила не менее 5000 т. т., по пику ацетилсалициловой кислоты не менее 7000 т. т., степень разделения всех пиков не ниже 2,5, фактор симметрии близок к единице. Относительное стандартное отклонение площади пика дигидрокверцетина 2,4%, ацетилсалициловой кислоты 1,2%. Все хроматографические параметры соответствуют современным фармакопейным требованиям.
6. Предложен селективный и воспроизводимый способ анализа композиции диквертина, аскорбиновой кислоты и b-каротина, как основы БАД каровертин и одноименного разрабатываемого препарата. Способ основан на предварительном фракционировании компонентов смеси методом твердофазной экстракции и последующем анализе фракций методом ВЭЖХ (аскорбиновая кислота и компоненты диквертина) и УФ-спектрофотометрии (b-каротин).
7. Изучен комплекс валидационных характеристик предложенных методов анализа и стандартизации препаратов диквертин, асковертин и саливертин в субстанции и лек. формах. По результатам валидации установлено, что все разработанные методики являются специфичными для определения содержания действующих веществ, характеризуются корректной точностью (правильностью) и воспроизводимостью, линейной зависимостью в аналитической области ± 30% по отношению к заявленному содержанию действующих веществ в препарате, что позволяет использовать их для достоверной оценки качества препаратов.
8. Показано, что унифицированные методики анализа препаратов диквертин, асковертин и саливертин можно использовать для сквозной стандартизации препаратов по фармакопейным показателям подлинность, родственные соединения, посторонние примеси, растворение, однородность дозирования, количественное определение. Методики включены в нормативную документацию по стандартизации и контролю качества вышеперечисленных препаратов.
Список работ опубликованных по теме диссертации
1. , , и др. Разработка новой биологически активной добавки к пище «Антоксид». // Материалы IV международного съезда «Актуальные проблемы создания новых лекарственных препаратов природного происхождения». Великий Новгород. – 2001. – С. 210–216.
2. , , и др. Разработка методов анализа смеси диквертина и ацетилсалициловой кислоты – действующей основы нового комплексного препарата // Материалы VI международного съезда «Актуальные проблемы создания новых лекарственных препаратов природного происхождения». Санкт-Петербург. – 2002. – С. 184–186.
3. , , Тюкавкина анализ композиции диквертина и аскорбиновой кислоты. // Материалы VI Симпозиума по фенольным соединениям. М., 2004. – С. 106.
4. , , и др. Валидация методики анализа препарата «Асковертин» // Фармация№ 3. - С. 11-14.
5. , , и др. Валидность методики определения компонентов препарата «Саливертин» // Фармация№ 3. - С. 11-14.
6. , , и др. Стандартизация композиции аскорбиновой кислоты, диквертина и b-каротина // Материалы XII Российского нац. конгр. «Человек и лекарство». М., 2005. – С. 702.
7. , , и др. Разработка фармакопейного хроматографического метода анализа нового отечественного препарата саливертин // Материалы IX междунар. съезда «Актуальные проблемы создания новых лекарственных препаратов природного происхождения». Санкт-Петербург, 2005. – С. 556–559.
8. , , Тюкавкина примеси салициловой кислоты в новом препарате «Саливертин» методом ВЭЖХ // Микроэлементы в медицине. – 2005. – Т. 6. Вып. 3. – С. 90–93.
9. , , и др. Физико-химическая характеристика дигидрокверцетина как стандартного образца // Материалы Х междунар. съезда «Актуальные проблемы создания новых лекарственных препаратов природного происхождения». Санкт-Петербург, 2006. – С. 338–342.
