

Партнерка на США и Канаду по недвижимости, выплаты в крипто
- 30% recurring commission
- Выплаты в USDT
- Вывод каждую неделю
- Комиссия до 5 лет за каждого referral
ЛЕКЦИЯ 4. КЛАССИФИКАЦИЯ МУТАЦИЙ
, ИЦиГ СО РАН и ФЕН НГУ, Новосибирск, 2012 г.
4.1. Классификация мутаций по их молекулярной природе.
4.1.1. Точковые мутации
С точки зрения традиционного построения курсов генетики, мы с вами непростительно долго задержались на обзоре генетических понятий и тех молекулярных явлений, которые за ними стоят, и до сих пор не перешли к тому, что составляет суть классической генетики – генетическому анализу. Однако все же необходимо завершить понятийно-терминологический обзор, рассмотрев классификацию мутаций. В полной мере эта тема может быть раскрыта только одновременно с рассмотрением взаимодействия генов. Кроме того, оставим пока в стороне вопросы мутагенеза, то есть как, почему, в каком количестве мутации возникают и как их можно сосчитать - и ограничимся лишь его результатами, то есть самими мутациями. Начнем с классификации, основанной на характере молекулярного изменения, произошедшего в генетическом материале, с учетом его смысла с точки зрения генетического кода.
Прежде всего, выделяют точковые мутации. Точковые мутации в узком смысле представляют собой замены одних нуклеотидов на другие. Среди точковых мутаций выделяют:
- транзиции: нуклеотидные замены с сохранением класса азотистого основания, которые меняют один пурин на другой – тимин на цитозин или наоборот - или один пиримидин на другой – аденин на гуанин или наоборот.
- трансверзии: нуклеотидные замены с изменением класса азотистого основания.
Различение транзиций и трансверзий имеет значение постольку, поскольку транзиции в третьих позициях многих кодонов (а у некоторых кодонов – и во вторых) обычно не приводят к аминокислотным заменам. Это обстоятельство связано с тем, что генетический код, «принятый» у ныне существующих организмов, не так уж сильно - всего лишь набором уникальных исключений – отличается от так называемого идеального кода, при котором аминокислота определялась первыми двумя и всего лишь типом (пуриновый или пиримидиновый) третьего нуклеотида в кодоне; и даже количество уникальных тРНК варьирует в разных группах вокруг цифры 32, то есть при спаривании их антикодонов с кодоном оказывается неважным, какой именно пурин или пиримидин находится в третьей позиции.
Таким образом мы с вами подошли к существованию синонимичных и несинонимичных нуклеотидных замен – первые не приводят к изменению смысла кодона (кодирует определенную аминокислоту или является стоп-кодоном), вторые приводят. Данная бинарная классификация применяется только к кодирующим участкам ДНК – экзонам; нуклеотидные замены в некодирующих участках называть «синонимичными» не принято.
В свою очередь, несинонимичные нуклеотидные замены, меняющие смысл кодона в рамках генетического кода, делятся на миссенс-мутации – нуклеотидные замены, приводящие к замене одной аминокислоты на другую, и нонсенс-мутации – нуклеотидные замены, превращающие кодон, кодирующий аминокислоту, в стоп-кодон. Существование термина «нонсенс-мутации» имеет скорее исторические корни и не особенно оправдано – по крайней мере мутации, превращающие стоп-кодон в кодирующий, а также аналогичные мутации, затрагивающие старт-кодон, в отдельные классы не выделяются, что непоследовательно.
Миссенс-мутации в свою очередь делятся на консервативные – мутации, приводящие к заменам аминокислот в пределах своего класса, и неконсервативные – мутации, приводящие к изменению класса аминокислоты, кодируемой кодоном. Легко понятно, что консервативные замены в среднем меньше сказываются на функциях белкового продукта, чем неконсервативные, хотя, естественно, имеются и исключения.
Примеров точковых мутаций масса; поскольку в аудитории половина медиков, давайте возьмем пример из генетики человека. Впечатляющий иллюстративный материал для обзора различных типов мутаций являют гены глобинов человека. На самом деле на современном этапе развития генетики такой материал может дать едва ли не любой ген любого из основных генетических объектов.
Ген β-глобина обозначается HBB и расположен в хромосоме 11. Из 146 его кодонов, точковые мутации, общим числом 335 по состоянию на 2010 г были известны для как минимум для 138. На следующем рисунке приведен пример синонимичной замены А>С в гене (вверху) и случай неконсервативной миссенс-мутации, сменившей гистидин на лейцин (неконсервативная замена!), которая была выявлена у одной совершенно здоровой семьи из Граца (внизу).
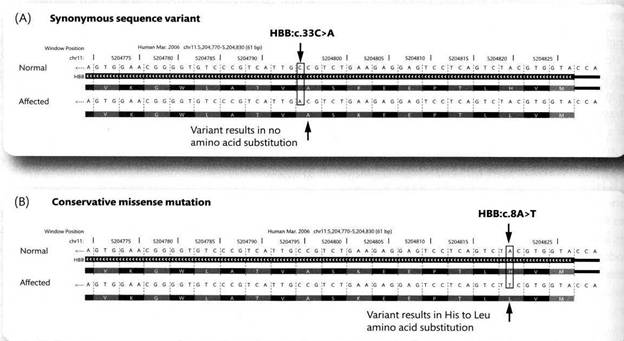
Напоминаю, что принято приводить нуклеотидные последовательности кодирующей цепи ДНК, то есть цепи, комплементарной матричной цепи – той цепи, по которой строится мРНК. Последовательность мРНК совпадает с последовательностью кодирующей части кодирующей цепи с той разницей, что вместо цитозина в ней стоит урацил. Обращаю внимание, что на этой и последующих иллюстрациях, касающихся гена β-глобина HBB, (позаимствованных из книги J. C. Knight “Human Genetics Diversity”), 5’-конец кодирующей цепи предполагается справа, транскрипция и трансляция предполагается идущей справа налево. Направление «чтения» связано с тем, что гены в этой книге привязаны к сквозной нумерации нуклеотидных позиций на хромосоме (мелкие цифры сверху). Эта ориентация непривычна – мы вынуждены читать ген справа налево, но нужно привыкать, что может встретиться и такое. На всякий случай напоминаю также, что гены (пардон, транскрибируемые последовательности) в хромосоме в целом ориентированы «как попало» - некоторые параллельно, некоторые антипараллельно, хотя каждый ген в своем локусе ориентирован строго определенным образом и все экзоны одного гена ориентированы параллельно. Поэтому одна и та же из двух цепей ДНК в своих кодирующих участках бывает то матричной, то кодирующей (но только какой-нибудь одной во всех экзонах одного и того же гена); соотсветственно, одни гены будут «читаться» справа налево, другие слева направо.
Выше мы уже рассмотрели серповидно-клеточную анемию, которая связана с неконсервативной несинонимичной заменой в том же самом гене, результате чего полярная аминокислота глутамат заменилась на алифатическую – валин, и белок с такой заменой вызывает патологические последствия. Кстати, в результате другой нуклеотидной замены тот же остаток глутамата заменяется на остаток лизина – также с гидрофильным заряженным радикалом, но с противоположным знаком заряда. В результате образуется так называемый гемоглобин С. У гетерозигот по такому аллелю он составляет 28-44% всего гемоглобина, в этом отношении мутация полудоминантна. Однако на здоровье человека это обстоятельство не сказывается, в этом отношении мутация рецессивна. У гомозигот весь гемоглобин оказывается гемоглобином С, и это нарушает нормальную пластичность эритроцитов и приводит к умеренной анемии. Данная мутация в гетерозиготе также дает определенную устойчивость к малярии и выглядит более приемлемым решением проблемы малярии, чем та, которая связана с сепповидно-клеточной анемией. Она широко распространена в Западной Африке, а также у происходящего оттуда негритянского населения Северной Америки.
При серповидно-клеточной анемии нарушено качество молекулы β-глобина, при сохранении ее количества. Если же какой-то из двух глобинов из состава гемоглобина, синтезируется в недостаточном количестве, это приводит к наследственным заболеваниям, известным под общим названиям «талассемии». В случае недостатка α-глобина, молекулы β-глобина формируют нефункциональный тетрамер. В случае недостатка β-глобина, избыточные молекулы α-глобина связываются с мембраной эритроцита. Если дисбаланс между глобинами велик, то за счет этого созревание многих эритроцитов нарушается и они разрушаются еще в красном костном мозге, а те, которые все-таки входят в кровяное русло, имеют дефект мембраны и довольно быстро разрушаются в селезенке. Большинство мутаций, приводящих к талассемиям, полудоминантны с точки зрения количества соответствующих глобинов в эритроцитах. С точки зрения патологии их можно назвать рецессивными, так как гетерозиготы как правило не имеют проблем со здоровьем. Существует два паралогичных гена α-глобина, HBA1 и HBA2 (хромосома 16) и лишь один ген β-глобина, поэтому потеря функции одним из генов α-глобина не столь сильно сказывается на организме, как потеря функции гена β-глобина. В гомозиготе по мутации, приводящей к потере функции одного из двух генов α-глобина, формируется так называемая «малая талассемия», которую можно установить лишь по слегка уменьшенному объему эритроцитов; она практически не сказывается на жизни пациентов, разве что анемия может развиться при болезни или беременности. В гомозиготе мутация, связанная отсутствием или резким сокращением количества β-глобина, вызывает так называемую «большую талассемию», для которой характерна анемия, повреждение костей, увеличенная селезенка и большое количество несвязанного железа в организме. Без периодической гемотрансфузии больные не доживают до 20 лет.
Вы знаете, откуда произошло название «талассемия»? «Θάλασσα» по-гречески – море (слово неиндоевропейского происхождения; судя по всему, греческий язык формировался настолько далеко от моря, где-то в степях Восточной Европы, что его носители потеряли свое индоевропейское «маре» и вынуждены были заимствовать слово у «загадочных пеласгов» – догреческого населения нынешнего греческого мира). Дело в том, что концентрация β-талассемии велика в Средизмноморье, особенно в восточном, а именно в Греции, в Эгейском регионе Турции (также изначально бывшем частью греческого мира) и на островах Средиземного моря, где в некоторых районах носителями мутации в гетерозиготе является до 20% населения. Считается, что в мире живет 60-80 млн носителей β-талассемии. В настоящее время выявлено около 200 различных (!) мутаций, приводящих к β-талассемии, и подавляющее большинство из них – точковые мутации, то есть замены нуклеотидов, реже выпадения 1-2 нуклеотидов.
В чем же причина такой концентрации мутаций по гену β-глобина в Средиземноморье? Распространенность талассемий в целом коррелирует с распространенностью малярии, что, как и в случаях с серповидно-клеточной анемией, позволило и в данном случае выдвинуть гипотезу о сбалансированном полиморфизме, основанном на сверхдоминировании – преимуществе гетерозигот, которые предполагаются более устойчивыми к малярии.
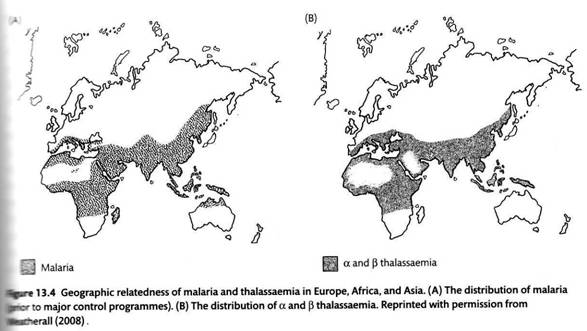
Данные, подтверждающие устойчивость к малярии гетерозигот по талассемичеким мутациям, действительно были получены, но в основном касательно α-талассемий, причем разные мутации в гетерозиготе обеспечивают разную устойчивость к разным видам малярии. Во многих районах Юго-Восточной Азии те же самые 20% населения являются носителями мутаций, имеющих отношение к α-талассемии.
Так вот, средиземноморская талассемия в основном связана с двумя наиболее распространенными аллелями. Одна из них преобладает в западных районах Средиземноморья - Алжир, Тунис, Испания, Португалия, Кипр, Сардиния - и связана с нонсенс-заменой C на T в 39 позиции кодирующей части локуса (обозначено CD 39 (C>T), здесь CD означает Coding Domain). Самая первая нонсенс-мутация, выявленная у человека, также была связана с β-талассемией, распространенной в Китае, и была связана с заменой A>T в 52 позиции, которая превратила лизиновый кодон AAG в стоп-кодон TAG, как показано на следующем рисунке:
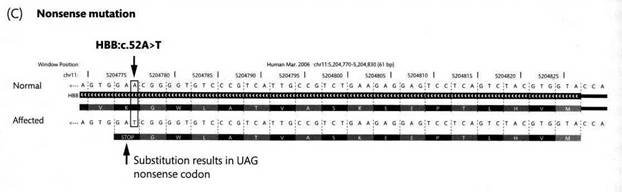
Скажите, что должно происходить в клетке, если в каком-то гене возникает нонсенс-мутация? Ее результатом должна быть преждевременная терминация трансляции. В результате в клетке должен бы синтезироваться укороченный и скорее всего нефункциональный полипептид, скорее всего в тех же молярных количествах, в каких должен был бы присутствовать нормальный. Однако еще в 80е годы ученых, в частности исследователей талассемий и обсуждаемой ниже болезни Тея-Сакса, весьма смущал тот факт, что они не обнаруживают в таких клетках не только укороченного полипептида в каких-либо количествах, но даже и соответствующей мРНК. Как недавно выяснилось, у эукариот – от дрожжей до человека – существует специальным механизм, nonsense-mediated mRNA decay (NMD). (Не уверен, что кто-нибудь уже умудрился перевести это на русский язык.). Этот механизм уничтожает процессированную мРНК если открытая рамка считывания обрывается ранее чем за 50-55 нуклеотидов до последнего экзон-экзонного стыка. Дело в том, что во время процессинга с областью мРНК 20-24 нуклеотида до (с 5’-стороны от) каждого экзонн-экзонного стыка связываются белки, входящие в так называемый exon junction complexes. Всякая мРНК претерпевает пионерный раунд трансляции в ядре (!), в ходе которого рибосома при своем движении относительно мРНК снимает эти белки. Затем мРНК транспортируется в цитозоль. При наличии нонсенс-мутации, компоненты EJC с 3’ стороны от нее остаются не снятыми рибосомой и опознаются в цитозоле определенным белком, который инициирует дергадацию такой мРНК.
Существуют классы точковых мутаций, которые имеют критическое значение для молекулярной функции гена, но эти классы не имеют столь красивых названий. Это исчезновение и возникновение сайтов сплайсинга, а также альтенативных промоторов.
Канонический сплайсинг проходит по консервативным (но не абсолютно) последовательностям, между GТ на 5’конце (сайт-донор) и AG, перед которым - много пиримидинов (C и U) - на 3’конце (сайт-акцептор). Исчезновение сайта сплайсинга может привести:
- к включению интрона в зрелую мРНК, что может привести к трансляции бессмысленной интронной последовательности, появлению в ней стоп-кодона, сдвигу рамки считывания (об этом ниже),
- к выпадению экзона, если сплайсинг пойдет до конца следующего интрона.
Мутация может уменьшить специфичность и точность сплайсинга – тогда в экзоне может добавится или исчезнет несколько нуклеотидов или появится вариант альтернативного сплайсинга.
Важность таких мутаций легко понять, если учесть, что около 80% генов человека свойственен не просто сплайсинг, а альтернативный сплайсинг. Какой именно вариант сплайсинга будет иметь место в данной клетке зависит от многих факторов, во многом на этом построена регуляция генных сетей, и любая мутация, затрагивающая сайт сплайсинга, нарушит существующую регуляторную систему (или создаст предпосылки для ее нарушения)ы. Для сравнения, у нематоды альтернативный сплайсинг проявляют всего около 15% генов.
Мы с вами говорили о β-талассемии, имеющей большие частоты среди населения Средиземноморья и упомянули один аллель, связанный с нонсенс-мутацией. Второй наиболее распространенный аллель связан с заменой G>A в 110й позиции первого интрона, и нарушает нормальное течение сплайсинга в ходе процессинга мРНК (обозначается IVS I 110 (G>A), аббревиатура – от interveining sequence), что приводит к резкому снижению количества, но не полному исчезновению β-глобина. Этот аллель преобладает в восточных районах Средиземноморья – в Югославии, Греции, Турции, с максимумом на Кипре. Однако в Средиземноморье много и других аллелей. Лучше всего исследован этот вопрос в Италии. На одной только Сицилии у 5,9 % населения имеются проблемы, связанные с недостатком β-глобина. Там выявлено 15 разных (!) мутаций гена HBB, приводящих к β-талассемии (против 6 в нескольких генах α-глобинов), а во всей Италии – 21 аллель. Эти аллели перечислены на следующей таблице, где показана также их локализация и молекулярная природа. Обратите внимание на традиционное для исследований талассемии, но нетрадиционное для генетики обозначение фенотипов: β+ означает не нормальный фенотип, а присутствие у гомозигот β-глобина хотя бы в каком-то количестве, β0 указывает на его полное отсутствие. Вторая колонка цифр показывет количество индивидуальных аллелей, разных или одинаковых, выявленных в исследовании 2005 г. (по вполне понятным причинам один пациент может поставить исследователям от одного до двух мутантных аллелей), третья – процентная встречаемость данного аллеля среди всех исследованных случаев талассемии. Частоты аллелей в популяции не приведены, поскольку гетерозиготы с «диким типом» обычно нормальны и не попадают в поле зрения медицинской генетики.
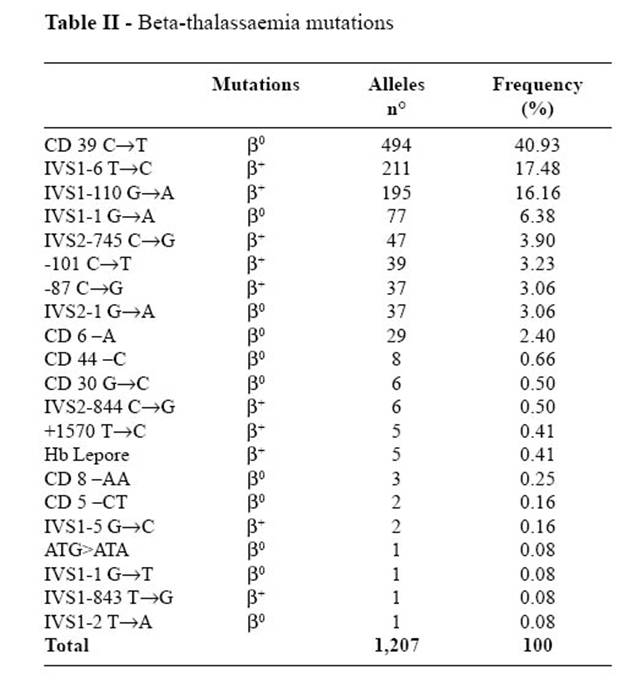
Заметим два обстоятельства:
1. - 90% случаев β-талассемии в Италии связано всего с четырьмя аллелями,
2. - одного этого обстоятельства достаточно, чтобы в поле зрения исследователей попадали индивидуумы с самым разным генотипом, в частности такие, у которых разные мутантные аллели находятся в компаунде, что может приводить к особенностям талассемического фенотипа.
Примечательна замена G>А в первой позиции 2го интрона (IVS II), уничтожающая сайт-донор. В результате, вместо нормального сплайсинга имеют место два варианта альтернативного сплайсинга – оба аномальные. В одном из них второй экзон полностью выпадает, в другом активируется криптический сайт-донор, в результате чего первый экзон удлиняется на 19 аминокислотных остатков. В гомозиготе по такой мутации развивается тяжелая β0 талассемия.

Мутация может не только уничтожить, но и создать сайт сплайсинга. На следующем примере замена G>A в конце первого экзона создает альтернативный сайт-донорTG*GTА (немутантная последовательность TGGTG, хотя и имеет участок TGGT, сайтом-донором не является), в результате чего возникает альтернативный сплайсинг – некоторое количество мРНК сплайсируется теперь по нему, хотя в основном сплайсинг протекает нормально, по стандартному сайту сайту AG*GTT.
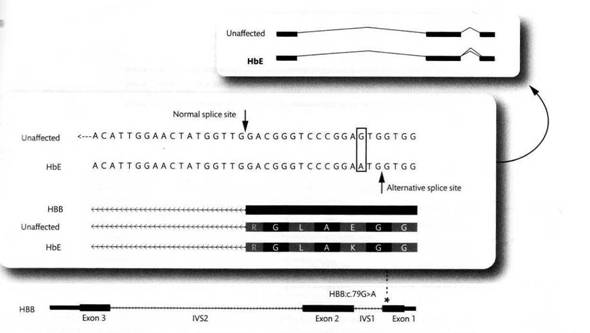
Кстати, данная замена при нормальном сплайсинге приводит также к замене глутамата на лизин, в результате чего образуется так называемый гемоглобин Е. Его носителем являются около 30 миллионов человек в Юго-Восточной Азии – естественно, также за счет некоторой устойчивости к малярии. Аллель, несущий данную мутацию, создает проблемы в компаунде с «талассемическими» аллелями, у таких гетерозигот развивается талассемия, требующая гемотрансфузии.
Переходим к следующему типу точковых мутаций, также не имеющему особого названия – мутации, затрагивающие регуляторные сайты. Посмотрите на аллель, занимающий в таблице почетное седьмое место. Кстати, он достигает высокой популяционной частоты в Болгарии – 2-6%! (Здесь уже имеется в виду не частота аллеля среди клинических случаев талассемии, а частота аллеля в общей популяции, то есть его доля среди всех вообще аллелей HBB, включая нормальные, имеющаяся у местных людей). Он также является точковой заменой С>G, но эта замена находится в позиции –87, то есть в 5’-направлении от начала кодирующей части, а именно в промоторе. Это довольно мягкий аллель, связанный с уменьшением уровня транскрипции и в гомозиготе приводящий к «промежуточной талассемии». Есть и другие аллели, приводящие к талассемиям, связанные с мутациями в 5’-некодирующей области гена HBB. Мы видим эффект точковой мутации в регуляторной, а не кодирующей области гена.
Наконец, в таблице у четвертого снизу аллеля мы видим и замену нуклеотида в самом старт-кодоне. Естественно, такая мутация исключает трансляцию β-глобина, и с ней, конечно же, связан фенотип β0.
Мы детально рассмотрели единственный и самый простой тип мутаций – нуклеотидные замены, которые не очень хорошо поддаются строгой классификации. Попробуем резюмировать раздел попыткой ее построения. С химической точки зрения нуклеотидные замены делятся на трансверзии и транзиции. С точки зрения места возникновения они делятся прежде всего на мутации в кодирующей и некодирующей ДНК. Мутации в кодирующей ДНК делятся на синонимичные и несинонимичные. Среди последних принято выделять на миссенс-мутации, меняющие одну кодируемую аминокислоту на другую; и нонсенс-мутации, меняющие кодон, кодирующий аминокислоту, на стоп-кодон. Здесь же можно упомянуть мутации, уничтожающие стоп-кодон и уничтожающие или создающие старт-кодон. Миссенс-мутации можно разделить, на приводящие к консервативным и неконсервативным заменам. Мутации в некодирующей ДНК можно разделить на нейтральные (незначащие) и на затрагивающие регуляторную область генов (и далее классифицировать какую именно и каким образом). Следует выделить также мутации, затрагивающие и не затрагивающие сайты сплайсинга; причем это деление независимо от всех предыдущих, так как они могут случаться в кодирующей и некодирующей ДНК и менять или нет смысл кодона. Мутации, затрагивающие сайты сплайсинга, можно разделить на влияющие на сайт-донор и на сайт-акцептор, на уничтожающие сайт сплайсинга или добавляющие его, на приводящие к альтернативному сплайсингу или исключающие альтернативный сплайсинг и, как мы увидим ниже, на приводящие и не приводящие к сдвигу рамки считывания.
4.1.2. Вставки и выпадения нуклеотидов, сдвиг рамки считывания.
Теперь посмотрим на аллель, стоящий на 9 месте. Это не нуклеотидная замена. Это вставка А (аденина) в позиции 6 кодирующей части. А на следующем рисунке показан случай выпадения одного нуклеотида, также А, в позиции 20 того же гена – в том же самом 6м кодоне, в котором мы наблюдали нуклеотидную замену, приведшую к серповидно-клеточной анемии. В результате уже 19й кодон оказывается стоп-кодоном. Оба случая приводят к β0 талассемии

Такие небольшие вставки (инсерции) и выпадения (делеции) иногда к относят к точковым мутациям, а иногда не относят (в английском языке для обозначения одновременно вставок и выпадений существует обобщающий термин indels). Нетрудно понять, что происходит в результате мутации, подобных упомянутой последней. Вставка или выпадение одного или некоторого, но не кратного трем, количества нуклеотидов, приводит к так называемому сдвигу рамки считывания. Возникающая проблема напрямую вытекает из таких свойств генетического кода, как непрерывность и смежность, а именно то, что триплеты нуклеотидов оказываются кодонами исключительно потому, что они «отсчитываются» по три от начала кодирующей части. Сдвиг такого отсчета на число, некратное трем, приведет к тому, что трансляционная машина будет воспринимать в качестве кодонов бессмысленные триплеты. Такие мутации называются мутации сдвига рамки считывания – frameshift mutations.
Каковы следствия такой мутации? Мы вынуждены предположить, что в 3’-направлении от нее полипептидная цепь будет составлена из случайных, бессмысленных аминокислотных остатков. Но кроме этого велика вероятность, что среди бессмысленных триплетов довольно скоро окажется стоп-кодон, то есть полипептидная цепь оборвется очень скоро – как это и произошло на примере, приведенном на предыдущем рисунке. Вероятность получить стоп-кодон в случайной последовательности нуклеотидов достаточно велика. И, между прочим, при расшифровке геномных сиквенсов кодирующие гены находят прежде всего в качестве открытых рамок считывания – протяженных участков, на которых не встречаются стоп-кодоны.
У человека существует так называемая болезнь Тея-Сакса (Tay-Sachs), которая проявляется с возраста около 6 месяцев и приводит к смерти в возрасте около 4 лет. Она заключается в слепоте, глухоте, невозможности нормально глотать, атрофии мышц, тяжелой умственной и физической недостаточности. Диагностическим является вишневое пятно на ретине – это видна центральная ямка, где ганглиозные клетки отсутствуют, которая сама по себе является нормальным явлением. Однако она контрастирует с окружающими ее ганглиозными клетками, которые окрашены светлее за счет накопления ганглиозидов - производных жирных кислот – во всех нервных клетках. Болезнь Тея-Сакса является следствием рецессивной мутации в гене HEXA α-субъединицы β-N-ацетил-D-гексоаминидазы, которая в норме присутствует в лизосомах и расщепляет липиды. (Β-гексоаминидаза состоит из двух субъединиц и небольшого белкового же кофактора – белка гликолипидного транспорта.) У гетерозигот активности достаточно для нормальной жизни, но можно выявлять недостаточную активность фермента в лейкоцитах и тем самым тестировать гетерозигот.
В некоторых популяциях частота мутации повышена. В частности, это имело место у евреев ашкенази, где частота болезни Тея-Сакса до середины прошлого века составляла около 1,5%, причем один из 27-30 человек являлся носителем. (Это примерно та же доля, что и носителей серповидно-клеточной анемии в Африке, и мы подсчитали, что ей должна соответствовать рождаемость гомозигот на уровне 3%, мы же имеем вдвое меньше. По-видимому, не все гомозиготы доживают до рождения). Это примерно в десять раз выше, чем у «окружающей» популяции. Около 70% случаев болезни Тея-Сакса у ашкенази вызвана единственным аллелем, имеющим вставку 4 нуклеотидов в экзоне 11, что приводит к терминации трансляции уже через 10 нуклеотидов за счет появления стоп-кодона (миссенс-мутация). Та же самая мутация обнаружена в популяции кейджн (cajuns, cadiens) Луизианы – потомков около 3 000 франкоканадцев, депортированных англичанами из Атлантической Канады в гг. (половина погибла в трюмах кораблей по дороге). Происхождение аллеля у кейджн удается проследить до одной супружеской пары-основательницы, жившей во Франции в том же XVIII веке, потомки которой по-видимому присоединились к кейджн уже после их поселения в Луизиане. (Остальные 25-30% случаев болезни Тея-Сакса у ашкенази связана с мутацией, нарушающей сплайсинг, что приводит к выпадению экзона 4.)
Так вот, рождение подобных младенцев уже в прошлом. Первый дешевый тест на выявление гетерозиготных носителей был разработан в Америке еще в 1969 г. В 1971 г. в Балтиморе и Вашингтоне 1800 ашкенази прошли добровольный скрининг. Всеобъемлющие и эффективные меры по пренатальному тестированию синдрома и выявлению носителей мутации в гетерозиготе, в Израиле, Канаде и США привели к тому, что к 2005 году «еврейская болезнь» была практически искоренена. Последний ребенок с синдромом родился в Израиле в 2003 г., в том же году в США родилось 10, но все они имели другие мутации, не выявлявшиеся при специфическом ПЦР-анализе. В Израиле скрининг и консультации бесплатны и охватывают все население, в частности, имеется программа, из которой пара может анонимно узнать, не является ли она несовместимой – то есть не являются ли молодой человек и девушка оба гетерозиготами по мутации. Успешные мероприятия по искоренению синдрома стимулировали аналогичные меры в отношении других наследственных заболеваний и сделали Израиль лидером и международным центром в области медицинской генетики.
Вообще же мутаций гена HEXA, приводящих к синдрому Тея-Сакса, обнаружено у людей самых разных национальностей множество – к 2000 году их было известно около сотни.
Хочу сделать одно лирическое отступление. Я очень прошу не делать выводов об антисемитизме на основе одного только упоминания слова «еврей». Неверным было бы также первое впечатление, что для евреев особенно характерны наследственные заболевания – другие популяции имеют высокие частоты других наследственных заболеваний, но об этом не так хорошо известно. Изучение еврейских популяций дало очень много для популяционной генетики человека, и это происходит, во-первых, из-за высокоразвитой медицины и медицинской генетики в Израиле, где скринингом наследственных заболеваний охвачено все население - и это неудивительно, учитывая тот факт, что медицина искони была одной из традиционных еврейских профессий. Во-вторых еврейский народ имеет весьма интересную и богатую историю, что всегда привлекало внимание исследователей. В частности, упомянутые евреи Ашкенази (происходит от еврейского названия средневековых германских земель – Ашкеназ – как места расселения потомков Аскеназа, внука Иафета; и это, в частности, те евреи, которые живут в России), а именно потомки евреев, которые пришли вместе с арабами на Пиренейский полуостров в качестве носителей «интеллигентных» профессий и были изгнаны оттуда во время Реконкисты, а также латиноязычных евреев Галлии. Они в основном поселились в Германии, где жили в X-XIII веках и приобрели язык идиш (вариант средненемецкого, обогащенный заимствованием из иврита) и характерную традиционную манеру одеваться, которую можно наблюдать у религиозных людей в Израиле и которая совсем не библейская, а воспроизводит одеяние средневековых немецких горожан. Религиозная нетерпимость в Европе росла и подогревалась движением крестоносцев, воевавших со славянским востоком. На этом фоне, в XIII веке польский король Казимир III Великий пригласил немецких евреев в Польшу, как носителей тех самых необходимых ему «интеллигентных» профессий. Первоначальная популяция составляла около 25 тысяч, однако впоследствии она выросла до нескольких миллионов. Оттуда они заселили также Украину и Белоруссию, входивших в состав Польского королевства и Речи Посполитой. Таким образом, эта популяция прошла через несколько периодов очень небольшой численности, так что в результате эффекта бутылочного горлышка некоторые аллели вне зависимости от своего эффекта на фенотип могли достичь высокой численности в силу случайных причин (так называемого генетического дрейфа). Но и это еще не все. Вы знаете, американский штат Юта славится своим мракобесием – там, кажется, запрещено преподавать дарвинизм в школах. Но вот группа генетиков человека под руководством Кохрана из университете Солт-Лэйк-Сити отличается бескомпромиссным дарвинистическим подходом применительно к человеку. Они обратили внимание, что большинство наследственных заболеваний в популяции ашкенази, в частности и болезнь Тея-Сакса, имеют отношение к одной странице учебника биохимии, посвященной метаболизму липидов, в частности сфинголипидов. Они связали такое обстоятельство с тем, что во время быстрого роста численности популяции ашкенази в Польше имел место естественный (!) отбор на повышение интеллекта, поскольку количество детей якобы реально зависело от успеха в свойственных им интеллектуальных профессиях – ситуация, не частая у современных людей. Заметим, что именно к ашкенази относятся все те многочисленные евреи, которые внесли столь большой вклад в западные культуру и науку. Возможно, в результате такого отбора на интеллект и зафиксировались мутации, вредные в гомозиготе, но повышающие эффективность работы мозга, возможно через большее развитие дендритной системы нейронов, в гетерозиготе. Надо сказать, что еще в 80е годы, когда было обнаружено, что высокая частота этого наследственного заболевания у ашкенази связана как минимум с двумя разными мутациями, была выдвинута гипотеза, что эта высокая частота вызвана каким-то селективным преимуществом гетерозигот.
Все эти факты, с одной стороны, безумно интересны, с другой – совершенно исключают какие-то оценочные тенденции относительно того, какой народ «лучше», а какой «хуже». Как, например, и такой факт, что до десяти процентов ядерного генома современные люди, судя по всему, приобрели от неандертальцев в результате «скрещиваний» с ним во время своей экспансии из Африки (то есть много позже их дивергенции), но это не касается коренного населения Африки, которое осталось свободным от заимствований. Спрашивается – «хуже» европейцы африканцев, если содержат в геноме последовательности подвида, проигравшего в конкуренции современному подвиду, или лучше, если пока что выигрывают в могуществе у самих африканцев?
Но вернемся к мутациям сдвига рамки считывания. На следующем рисунке приведены последствия выпадения пяти нуклеотидов в начале первого интрона в гене HBA2. (Обращаю внимание, что здесь 5’конец находится слева, ген следует читать привычным образом слева направо). Казалось бы, выпадение нуклеотидов в некодирующей части не должно влиять на белок. Однако при этом опять-таки пострадал сайт-донор сплайсинга AGGT. В результате в качестве такового стал использоваться «криптический» сайт GGGT, уже имевшийся в кодирующей части. Любопытно, что этот не совсем канонический сайт в данном случае работает исправно – никакого альтернативного сплайсинга или непроцессированной мРНК не наблюдается. Однако обратим внимание, что нормальный сплайсинг проходит между вторым и третьим нуклеотидом в триплете, который находится в фазе кодирующей части первого экзона, тогда как новый сайт расположен между первым и вторым нуклеотидом кодона. В результате второй и третий экзоны оказываются не в фазе и мы снова имеем сдвиг рамки считывания.
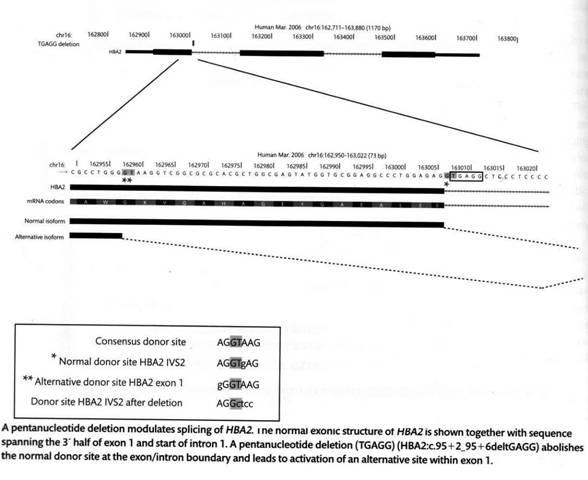
Альтернативный сплайсинг, возникающий при обсуждавшейся выше мутации, приводящей к гемоглобину Е, также происходит не после второго нуклеотида кодона, как в норме, а после первого, что приводит к сдвигу рамки считывания во втором и третьем экзонах. Именно с этим связано не только изменение качества, но и уменьшение количества β-глобина, так как мРНК, несущая сдвиг, содержит стоп-кодон и удаляется механизмом NMRD.
Мутации сдвига рамки, если они происходят в конце гена, могут приводить и к удлинению белкового продукта, поскольку стоп-кодон оказывается не в фазе. Инсерция двух нуклеотидов, СА, в конце гена HBB удлинает его на 11 аминокислот, так как уничтожает стоп-кодон UAA в 147 позиции кодирующей части. Это приводит к усилению сродства гемоглобина к кислороду и увеличение количества эритроцитов в крови.
А так называемый α-глобин Констант-Спринг (HBA-CS) содержит лишние 31 аминокислотных остатков. (Он назван так, поскольку обнаружен у семьи китайского происхождения в ямайском округе Констант Спринг.) Соответствующая ему мРНК нестабильна и в значительной степени деградирует до трансляции, белковый продукт также нестабилен. В результате мутация также проявляет талассемический фенотип.
Между прочим, мутации сдвига рамки считывания, особенно в семействах генов-паралогов, могут быть основой быстрого способа эволюции белковых последовательностей, поскольку порождают отрезки случайных последовательностей аминокислотных остатков, которые случайно же могут иметь какую-то полезную молекулярную функцию, тогда как «старую» молекулярную функцию продолжат выполнять оставшиеся паралоги. Возникал предположение, что возникшая у бактерий сточных водоемов фабрики по производству нейлона в Японии нейлоназа возникла именно таким путем, однако это предположение не подтвердилось впоследствии. В то же время, в одной статье 2007 года утверждается, что сравнительный анализ человеческого и мышиного генома выявил у человека 470 и у мыши 108 случаев возникновения новых участков белковых последовательностей за счет сдвига рамки считывания: такую интерпретацию предполагали различия в кодирующей части ортологичных генов у этих двух объектов, когда один из них имел «нормальный» конец гена, а другой – «сдвинутый».
Не все выпадения и вставки нуклеотидов приводят к сдвигу рамки считывания. Так, 2/3 случаев уже упоминавшегосся выше муковисцидоза (cystic fibrosis) в мире и около 90% в США связаны с одной и той же делецией трех нуклеотидов в гене CFTR (хромосома 7), приводящая к выпадению одного фенилаланина в позиции 508 (!) белка, являющемся АТФ-зависимым трансмембранным каналом ионов хлора и тиоцианата в эпителиальных клетках, в результате чего он не может принять нормальную третичную структуру. Заметим, что всего у человека описано около 1400 различных мутаций, приводящих к муковисцидозу. Этот показывает нам:
- до какой степени в настоящее время развита медицинская генетика – к сожалению, пока только за рубежом.
- какой хорошей мишенью для мутаций является протяженный ген: ген CFTR, вместе с интронами, занимает около пар оснований, а его белковый продукт состоит из 1480 аминокислотных остатков.
- сколь осторожным нужно быть в суждениях о том, с одинаковыми или разными аллелями мы имеем дело при их одинаковом фенотипическом проявлении.
4.1.3. Делеции и инсерции
Выше мы уже рассмотрели делеции и инсерции очень небольших отрезков ДНК, важных не столько в качестве потери или приобретения имеющегося в них генетического содержания, сколько в нарушении правильного считывания триплетов, интерпретируемых в качестве кодонов, при трансляции. Такие мутации как правило называются мутациями сдвига рамки считывания. Выпадения или вставки протяженных отрезков ДНК, называются просто делециями и инсерциями.
В случае некоторых классов мобильных генетических элементов, о которых будет рассказано позднее, инсерция в одном месте связана с делецией в другом, то есть участок ДНК переместился с одного места на другое – такие события называются транспозицией, но транспозиция в чистом виде встречается не так часто. Исторически первыми были открыты мобильные генетические элементы, ведущие себя именно таким образом, после чего все весьма разнородные мобильные генетические элементы стали, довольно некорректно называть транспозонами. В том числе и такие элементы - ретротранспозоны, инсерция которых в одном месте не сопровождается делецией в другом месте, то есть, несмотря на название, транспозиция не имеет места. Такие элементы не перемещаются, а размножаются - с их РНК-транскрипта фермент обратная транскриптаза считывает новую ДНК-копия мобильного элемента, которая и встраивается в новое место генома.
Иногда какой-то участок ДНК в геноме удваивается, в таких случаях говорят о дупликации. Так, например, ведут себя вышеупомянутые ретротранспозоны. Если в результате дупликации две копии дуплицированной последовательности оказываются расположенными подряд голова к хвосту, это называется тандемной дупликацией.
Рассмотрим примеры, перекликающиеся с рассмотренными выше.
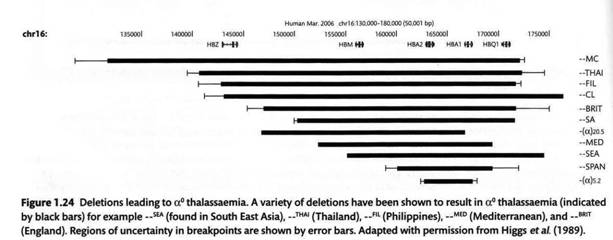
В отличие от аллелей, приводящих к β-талассемиям, большинство из которых связано с точковыми мутациями, большинство случаев α-талассемии связано с делециями. Тому есть две причины. Во-первых, α-глобин синтезируется двумя родственными паралогичными генами, HBA1 и HBA2, то есть в диплоидном ядре транскрипция в норме идет с четырех генов - две копии (полученные от отца и матери) HBA1 и две копии HBA2. Потеря функции одной и даже двух из них не сказывается на здоровье, талассемия обычно регистрируется, а пациент попадает в поле зрения генетика, только если оба гена выведены из строя, что легче сделать одной делецией, чем двумя точковыми мутациями. Во-вторых, оба паралога произошли в результате дупликации не так давно (порядка десяти миллионов лет назад) и их последовательности достаточно сходны. Более того, неподалеку находятся другие гены глобинов. Все они с низкой вероятностью могут «незаконно» выравниваться друг относительно друга в профазе мейоза, и в такой ситуации между генами возможен «незаконный» кроссинговер, который правильнее называть неаллельной гомологической рекомбинацией или неравным кроссинговером. В результате один гомолог приобретает лишние копии генов, а другой теряет какие-то копии.
На следующем рисунке схематически показано распроложение в глобиновом кластере делеций, вызывающих α-талассемии. Видно, что большинство из них перекрывают оба гена α-глобина и затрагивают участки хромосомы от одного гена до другого.
Зарегистрированы и носители дупликаций глобиновых генов, у которых в одной хромосоме находится не два, а три гена α-глобинов, так, в одной семье из Уэльса отец и два его сына имели три копии в одном гомологе и две (норма) в другом. У них наблюдался 20% превышение количества мРНК α-глобинов над мРНК β-глобина, что никак не сказывалось на «медицинском» фенотипе.
А на следующем рисунке показана схема делеций, которые вызывают α-талассемию, но не затрагивают самих генов α-глобинов. Зато все они затрагивают консервативные на межвидовом уровне последовательности MCS-R1 и MCS-R2 далеко от обоих генов. Эти последовательности совпадают с участками гиперчувствительности к ДНКазе I за счет того, что имеют более рыхлую структуру хроматина. Это и есть энхаснеры глобиновых генов, способствующие связыванию РНК-полимеразы и транс-активаторов транскрипции.

Выше мы упоминали болезнь Тея-Сакса. Частота этой болезни высока также в Квебеке, и хотя это также франкоязычная часть Канады, как и «Акадия», откуда были депортированы предки кейджн, мутация оказалась другой – делецией большого количества нуклеотидов. Когда болезнь Тея-Сакса обнаружили в Квебеке, относительно ее происхождения бытовала «теория еврейских торговцев пушниной», но потом выяснили природу мутации, а также то, что она восходит к семье, жившей в Южном Квебеке в XVII веке, то есть на столетие раньше, чем во Франции жила семья, давшая начало аллелю кейджн.
Иногда встречаются еще более экзотические мутации. Посмотрите еще раз на все тот же список аллелей, вызывающих β-талассемию в Италии, и обратите внимание на аллель четвертый с конца, носящий итальянское название «Лепоре». Для него не указан характер нуклеотидной замены. Дело в том, что это не совсем аллель гена HBB. Эта нуклеотидная последовательность представляет собой гибрид, между HBB и геном дельта-глобина HBD! Оба гена являются в известной степени паралогами, произошли в результате дупликации одного глобинового гена и их последовательности достаточно сходны. В результате произошел «незаконный же» кроссинговер, приведший к делеции около 7 тысяч пар оснований между этими генами и образованию гибридной последовательности глобинового гена. Эта последовательность унаследовала промотор от дельта-глобина, котороый слабо транскрибируется в эритроцитах, в результате β-глобина, пусть и не вполне полноценного, оказывается недостаточно и в гомозиготе развивается β-талассемия. Вот такие бывают делеции!
Более того! Среди гемоглобина Лепоре выявлены последовательности, в которых точка стыка дельта - и β-глобина варьируют, то есть такой гибридный ген возникал неоднократно: один происходит из Новой Гвинеи, другой – из Португалии и Бразилии, третий – из Испании и Италии. Найден и обратный гибрид, где начало гена происходит от β-глобина, а продолжение – от дельта-глобина!
Среди инсерции весьма распространены вставки мобильных генетических элементов, или транспозонов. Так например, классический аллель w у дрозофилы, гомозиготы по которому имеют непигментированные (белые) глаза, вызван вставкой не менее известного ретротранспозона copia. (Еще раз заметим, что ретротранспозоны способны встраиваться в геном, но неспособны из него выстригаться, то есть транпсозиция места не имеет.)
4.1.4. Хромосомные мутации
Наконец, к мутациям чисто формально можно отнести и изменения, касающиеся хромосом в целом, такие как транслокации, инверсии, или изменения числа хромосом, вплоть до удвоения (или иного кратного увеличения) всего генома. Их иногда даже называют хромосомными мутациями. Но в практике генетики слово мутация к ним обычно не применяется. Говорят конкретно о
- хромосомных перестройках - результатах разрывов хромосомной ДНК с последующим ее сшивания по оному,
- изменениях плоидности, то есть количества хромосом в ядре. Если оно кратно изменяется для всех хромосом гаплоидного набора, то говорят о гаплоидах (один набор), диплоидах, триплоидах, тетраплоидах и пр., а также в общем виде о полиплоидах. Если оно изменяетя некратно, по разному для разных негомологичных хромосом (при этом возникает дисбаланс в дозе генов, находящихся в негомологичных хромосомах), то говорят об анеуплоидах.
4.2. Классификации мутаций по месту и причине возникновения.
Мутации эукариот можно классифицировать также по тому, в каком геноме они происходят, то есть на ядерные в ядерном геноме и цитоплазматические – затрагивающие геномы митохондрий и хлоропластов.
Кроме того, по месту своего возникновения мутации делят на мутации в клетках зародышевого пути и в соматических клетках – естественно, в тех случаях, когда эти два пути разделены. Нельзя сказать, чтобы у первых было какое-либо специальное название, допустим, наследуемые мутации – в тех контекстах, когда о них заходит речь, такое уточнение как правило излишне. А вот вторые обычно называют соматическими мутациями. Им уделяется самое пристальное внимание хотя бы потому, что именно они являются причиной всех онкологических заболеваний.
Наконец, по характеру мутагенного фактора мутации делят на два больших класса – спонтанные и индуцированные. Ко вторым относятся мутации, вызванные преднамеренным воздействием на организмы мутагенных факторов, таких как химические мутагены или радиация (чаще всего используется гамма-излучение), а иногда также ультрафиолетовое облучение. Спонтанными же называют все мутации, которые происходят «сами собой» - как правило за счет ошибок ДНК-полимеразы, неправильного кроссинговера, воздействия вышеупомянутых мутагенных факторов в качестве естественных природных, а также под действием мобильных генетических элементов. Когда последние только открыли, одно время на них списывали большую часть спонтанных мутаций, но это оказалось преувеличением.
4.3. Классификация мутаций в отношении функции молекулярного продукта мутантных аллелей.
Предлагаю ознакомиться с классификацией мутаций с точки зрения того, что происходит с функцией молекулярного продукта соответствующего гена. Она была предложена Германом Меллером - одним из учеников Т. Моргана, фактически сооснователем генетики, особо интересовавшемся вопросами мутагенеза. Эта классификация чисто умозрительна, абстрактна и редко используется на практике. Тем не менее она очень полезна, что называется, для настройки мозгов, то есть для того, чтобы представлять весь спектр возможных последствий и не пропустить интересных случаев.
- аморфные мутации – молекулярная функция отсутствуют вовсе. Для мутация этого типа существует другой, уже знакомый Вам термин - нуль-аллель. Различают РНК-овый нуль-аллель – когда отсутствует транскрипт, включая сюда и делеции (их следовало бы назвать «ДНК-овый нуль-аллель», белковый нуль-аллель – когда отсутствует белковый продукт, и генетический нуль-аллель – когда продукт есть, но не выполняет свою молекулярную функцию.
- гипоморфные мутации – интенсивность функции меньше (в количественном или кинетическом смысле), чем в диком типе.
Оба предыдущих класса объединяются английским термином loss-of-fuction mutations.
- гиперморфные мутации – интенсивность функции больше, чем у дикого типа; соответствует английский термин gain-of-function mutations.
- антиморфные мутации – молекулярный продукт гена действует антагонистично к функции продукта дикого типа. Проиллюстрируем на гипотетической ситуации: допустим, продукт аллеля дикого типа является субстратом для какого-либо фермента, а продукт мутантного аллеля сохраняет способность связываться с его активным центром, но теряет способность вступать в данную ферментативную реакцию. Таким образом он блокирует активный центр за счет конкурентного ингибирования и тем самым из субстрата фермента превращается в эндогенный яд.
- неоморфные мутации – продукт приобретает новую молекулярную функцию и влияет на фенотип иным способом, чем аллель дикого типа.
4.4. Классифкация мутаций по действию на приспособленность.
Приспособленность – понятие из эволюционной биологии, означающее эволюционный, по сути репродуктивный, успех и будет детально рассмотрено в рамках соответствующего курса. Заметим здесь, что приспособленность включает в себя такие компоненты, как жизнеспособность, плодовитость и скорость индивидуального развития. (Нетрудно понять, что все три компоненты имеют отношение к репродуктивному успеху.) По влиянию на приспособленность выделяют мутации:
- летальные (lethal) – приводящие к смерти организма
- вредные (harmful or deleterious)
- полезные (beneficial)
- нейтральные (neutral)
Первый и последний класс требуют пояснений. В последний класс входят синонимичные, биохимические, не влияющие на функцию, а также часть морфологических. Исходя из компонентов приспособленности, мутации, не влияющие на жизнеспособность, но лишающие способности к размножению, следовало бы отнести к летальным – с эволюционной точки зрения их носители мертвы. Однако в практике генетики никто их не называет летальными.
4.5. Классификация мутаций по действию на фенотип.
В отличие от вышеприведенной, классификация по действию на разные аспекты фенотипа, несмотря на свою конкретность, не столь полезна, так как самоочевидна, тривиальна и не добавляет нам нового знания,:
- летальные мутации – носитель нежизнеспособен, фенотипа нет.
- морфологические мутации – влияют на морфологию организма.
- физиологические мутации – влияют на физиологию организма. Иногда выделяют класс поведенческий мутаций, но поведение – всего лишь одно из сложных физиологических проявлений, поэтому их скорее следует считать подклассом физиологических мутаций.
- биохимические мутации – их влияние можно выявить в биохимическом эксперименте, но они не влияют ни на одну физиологическую функцию. Сюда попадает масса мутаций, не сказывающихся на фенотипе в традиционном, а не молекулярном его понимании, то есть на «обычных» признаках организма. В частности, сюда электроморфы белков – их аллельные варианты (продукты разных аллелей одного локуса), различающиеся по электрофоретической подвижности. Хороший вопрос – куда следует относить мутации, влияющие на окраску? Логика заставляет считать их биохимическими мутациями. Однако их очень часто и довольно бездумно записывают в морфологические мутации, вопреки этимологии, указывающей на форму, понимая под морфологией все, что видно глазом.
- синонимичные мутации – вообще не выявляются в молекулярном продукте гена и могут быть обнаружены лишь при изучении своего материального носителя – соответствующего участка ДНК. Мы помним, что это тот курьезный случай, когда фенотип совпадает с генотипом.
Есть еще одна бинарная классификация мутаций по степени воздействия на фенотип, которая выглядит тривиальной, но имеет неожиданно большой смысл – это мутации с качественным эффектом и мутации с количественным эффектом. Суть этой классификации действительно тривиальна и следует из нашего рассмотрения качественных и количественных признаков. Мутации с качественным эффектом приводят к фенотипу, безошибочно отличающемуся от нормального. В том случае, если этот фенотип может быть измерен каким-то параметром, распределения значений этого параметра у нормальных особей и у особей, в которых проявляется данная мутация, не пересекаются. Эффект мутаций с количественным эффектом невелик и сопоставим с изменчивостью признака, привносимой изменчивостью внешней среды или даже с чисто случайной его изменчивостью. Как следствие, носителя такой мутации невозможно выявить по его фенотипу и генетический анализ оказывается возможным лишь методами генетики количественных признаков, оперирующими со многими мутациями подобного типа с неразличимым эффектом на фенотип. Данная «классификация» была бы недостойна упоминания здесь, если бы не одно важнейшее обстоятельство – мутации с небольшим количественным эффектом возникают гораздо чаще, чем мутации с качественным эффектом. А именно, частоты спонтанного возникновения первых имеют порядок 1/104на ген на поколение, а вторых 1/106, то есть мы имеем разницу в сто раз. Этот факт был обнаружен достаточно поздно, поскольку требовал изощренных методов анализа, которые сами по себе не менее интересны, чем причины этого феномена и чем его следствия. Возможно, у нас будет возможность рассмотреть методическую сторону этих исследований, сейчас вкратце коснемся причин и следствий.
Задним числом такую высокую частоту мутаций с небольшим количественным эффектом объяснить легко. Таким эффектом должны обладать мутации, влияющие не столько на кодирующие части генов, сколько на интенсивность их транскрипции. А мишенью для таких мутаций может быть весьма протяженная область ДНК вокруг кодирующей части, которая влияет на интенсивность транскрипции за счет цис-факторов - разнообразных сайтов связывания с белками-регуляторами, таких как энхансеры, и белками транскрипционной машины или каким-то иным образом.
Следствия из установленного факта высокой частоты мутаций с количественным эффектом также вполне серьезны. Организм представляет собой тонко отлаженную машину, построенную в ходе разворачивания тонко же отлаженной генетической программы. Любое изменение с гораздо большей вероятностью нарушит отладку и ухудшит машину в целом, чем улучшит ее (хотя все, что достигнуто эволюцией, достигнуто с помощью редких благоприятных мутаций). Поэтому среди мутаций с количественным эффектом, влияющих на важные интегральные признаки организма, на которые влияют множество генов, таких как размер тела, плодовитость, интеллект (там, где он имеется) и т. д. преобладают гипоморфные, слабо вредные мутации (slightly deleterious mutations). Следовательно, мутационный процесс сам по себе способствует постепенной деградации всей генетической системы и организма в целом. Единственной силой, противостоящей этой тенденции к «ухудшению качества» организмов, является естественный отбор. Если отвлечься от метафорического названия этого явления, то это означает следующее: если признак важен для приспособленности в данной среде, которая в свою очередь складывается из таких компонент, как выживаемость, плодовитость и скорость индивидуального развития, то автоматически появится тенденция исключения вредных мутаций из популяции. В результате в природе устанавливается динамическое равновесие между мутационным процессом, «ухудшающим» организмы за счет слабо вредных мутаций, и преимуществом во вкладе в следующее поколение, которое получают носители нормальных аллелей (и даже редких благоприятных мутаций). В условиях, когда какой-то признак перестает влиять на приспособленность, , он начинает деградировать, причем медленно и постепенно (ибо каждая мутация имеет весьма малый эффект). Мы можем наблюдать результат этого процесса на примере зрения, которое исчезает у пещерных организмов. Но в данном случае мы видим уже готовый результат длительной эволюции (не всегда чересчур длительной – известны случаи, когда слепые пещерные рыбы сохраняли способность скрещиватся в эксперименте со своими зрячими поверхностными родственниками, что позволило провести генетический анализ присутствия/отсутствия органов зрения). Еще один яркий пример такой деградации являют одомашненные виды животных, у которых признаки, не являющиеся хозяйственно-ценными, такие как объем мозга, развитие скелета и пр. – с очевидностью деградируют, причем эту деградацию можно констатировать археологически. (Сюда же, кстати, относятся и такие явления, как представляющиеся параллельными у разных видов «падение» ушей и появление пегостей в окраске.) Мы не можем наблюдать аналогичный процесс деградации интеллекта и физической силы у человека, поскольку находимся внутри очень медленного процесса, занимающего сотни и тысячи поколений. Но в том, что такая деградация человечества, незаметная, медленная и неуклонная, в целом имеет место, нет никаких сомнений, коль скоро количество оставленных потомков с какого-то момента развития человечества перестало коррелировать и с силой, и с интеллектом, а скорее наметилась обратная корреляция.
Вообще полезно бывает иногда увидеть мир таким, каков он есть. Американцы провели исследование репродуктивного успеха у людей в современном обществе, в котором выяснилось, что в этом отношении с огромным успехом лидировали несколько мужчин, являвшихся активными донорами в банки спермы. Поэтому советую молодым людям задуматься о таком способе подработки и достижения эволюционного успеха, поскольку там необходима молодость донора.
4.5. Закон гомологических рядов :
Вавилов сыграл огромную роль в развитии отечественной и мировой генетики, прежде всего заложив генетические основы селекции культурных растений. (Как и всякий выдающийся человек в СССР времен Сталина, он был арестован и, как и большинство из них, уничтожен. Он умер от дизентерии во время весьма продолжительного следствия. Его следователь впоследствии был всего лишь исключен из коммунистической партии, о чем очень переживал.) 4 июня 1920 г. на III селекционном съезде в Саратове он обнародовал положение, которое впоследствии стали называть Законом гомологических рядов в наследственной изменчивости . Вот его оригинальная формулировка: «Виды и роды, генетически близкие, характеризуются сходными рядами наследственной изменчивости с такой правильностью, что, зная ряд форм в пределах одного вида, можно предвидеть нахождение параллельных форм у других видов и родов.» Ставшими классическими примерами являлась сходная изменчивость культурных злаков, сходные наследственные варианты окраски у разных видов млекопитающих.
Это утверждение имело огромное практическое значение. Оно предполагало, что у разных видов культурных растений должны были существовать еще не известные к тому времени варианты изменчивости, имевшиеся у других видов. С целью обнаружения этих вариантов были организованы многочисленные экспедиции по всему миру, в частности в труднодоступные районы, в которых сохранилось древнее традиционное земледелие и традиционные культуры. И действительно, такие варианты как правило рано или поздно обнаруживались. Во вторых, этот закон в свое время имел и важное теоретическое значение и способствовал пониманию связи генетики и эволюции.
В то же время вызывает некоторое удивление, что многие в нашей стране называют это утверждение «законом» и превозносят вплоть до настоящего момента, видя в нем некое проявление одинаковой направленности эволюции у родственных форм, т. е. воспринимая его как проявление некоей общей эволюционной программы, несколько в духе «теории номогенеза» современника Вавилова . Последний ошибочно полагал, что подобно тому, как существует некая наследственная программа, управляющая онтогенезом – что становилось очевидным как раз в начале XX века, существует такая же наследственная программа, управляющая и филогенезом. На самом же деле сейчас «закон» гомологических рядов в наследственной изменчивости выглядит тривиальным. Речь идет не о направлении эволюции близких видов, а всего лишь об их мутациях, т. е. поломках генов. По сути, этот «закон» говорит лишь о том, что сходные организмы являются результатом развертывания сходной генетической программы их индивидуального развития. Программы индивидуального развития, а не эволюционных преобразований! Сходство программы предполагает и сходство ее изменчивости – в частности, в результате поломок каких-то ее элементов. (Кроме того, не исключено, что не все одинаковые мутации происходят у родственных видов независимо. Близкие виды могут иметь сходный полиморфизм просто потому, что они унаследовали этот полиморфизм от своего общего эволюционного предка.)
