
Партнерка на США и Канаду по недвижимости, выплаты в крипто
- 30% recurring commission
- Выплаты в USDT
- Вывод каждую неделю
- Комиссия до 5 лет за каждого referral
Рентгенофлуоресцентное определение содержаний рассеянных и некоторых основных элементов в алюмосиликатных горных породах с использованием спектрометра S4 EXPLORER
, *, ,
Институт земной коры СО РАН, Иркутск,
*Центральная аналитическая лаборатория Ботуобинской ГРЭ АК "АЛРОСА", Мирный, Республика САХА, Якутия
E-mail: *****@
1. ВВЕДЕНИЕ
Наибольшее число разрабатываемых в настоящее время месторождений алмаза относится к кимберлитам, поэтому эти породы являются объектом пристального внимания геологов, петрологов и минералогов [1]. Кимберлиты - особый тип магматических горных пород, в котором совмещены минералы, образовавшиеся в различных термодинамических условиях. Кимберлитовые брекчии содержат обломки осадочных пород чехла и кристаллических пород фундамента, а также ксенолиты глубинных пород мантии. Практическое значение кимберлитов определяется тем, что с этими породами связаны коренные месторождения алмаза. Среди алмазоносных ксенолитов по минеральному составу выделяются кристаллические биминеральные горные породы, обедненные кремнием и обогащенные алюминием, а также гранатовые перидотиты, обедненные кремнием и алюминием и обогащенные магнием и железом. При проведении поисковых работ по обнаружению алмазоносных кимберлитовых трубок необходимо располагать информацией об их геологическом расположении, механизме формирования, связи с другими магматическими породами глубинного залегания и сопутствующих минералах. Исследование элементного состава этих пород дает ответы на многие вопросы.
2. ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
За три года совместной работы Аналитического центра Института земной коры СО РАН г. Иркутска и Центральной аналитической лаборатории Ботуобинской геолого-разведывательной экспедиции (ЦАЛ БГРЭ) АК «АЛРОСА» разработаны методики количественного определения содержаний P2O5, S(общ.), Cl, K2O, TiO2, V, Cr, MnO, Co, Ni, Cu, Zn, Ga, Rb, Sr, Y, Zr, Nb, Mo, Ba, La, Ce, Nd, Pb, Th, U в различных геологических материалах.
2.1. Рентгеновский спектрометр S4 EXPLORER
Спектрометр S4 EXPLORER фирмы Bruker AXS (Германия) объединяет в себе аналитические возможности рентгеновского спектрометра с волновой дисперсией и малые габариты. Замкнутый охладительный контур смонтирован внутри прибора. Анализ элементов с малыми атомными номерами при помощи S4 EXPLORER базируется на применении многослойных кристаллов-анализаторов, грубых коллиматоров, отпаянного пропорционального счетчика Pro4 Super High Transmission и использовании расположенной близко к пробе рентгеновской трубки со сверхтонким (75 мкм) окном. Диапазон определяемых элементов - от бериллия до урана. Диапазон определяемых концентраций - от ppm до 100%. Спектрометр применим для анализа порошков, твёрдых тел, жидкостей, покрытий, паст, плёнок и др. Для уменьшения влияния шероховатостей поверхности образца на метрологические характеристики методики проба при измерении вращается со скоростью 30 об/мин. Спектрометр стандартно оснащён накопителем на 60 проб.
Функции и протекание процессов внутри спектрометра контролируют несколько микропроцессоров. Главный процессор координирует внутренние информационные потоки и осуществляет связь с внешним компьютером, обрабатывающим данные. Такой подход позволяет оптимизировать диагностические возможности и быстро локализовать неисправности.
Программное обеспечение SPECTRAplus спектрометра S4 EXPLORER поставляется вместе с библиотекой рентгеновских линий. Для приближённо-количественного анализа образцов неизвестного состава реализован способ фундаментальных параметров. Угловые позиции определяемых элементов были предварительно откалиброваны и откорректированы на заводе в г. Карлсруэ (Германия). Благодаря этой особенности можно производить анализ различных материалов по предварительно откалиброванным линиям. Используя эту программу можно при разработке методик для конкретных аналитических задач, выбирать условия и параметры измерения, производить измерение калибровочного массива, выбирать оптимальные калибровочные уравнения и осуществлять математическую обработку калибровочного массива стандартных образцов (СО) [2]. Данная возможность актуальна при анализе материалов с широким диапазоном варьирования состава матрицы (например, горных пород). Подробное описание технических особенностей спектрометра S4 EXPLORER можно найти в рекламных публикациях [3, 4].
2.2. Анализ спектров определяемых элементов
Рентгеновский спектр в диапазоне длин волн CeLb1 - BaLa1, измеренный для СО STM с использованием спектрометра S4 EXPLORER, представлен на рис. 1. Рисунок показывает существенное влияние наложения TiKa1-линии (0.2748 нм) на BaLa1-линию (0.2778 нм). Обычно в горных породах содержание TiO2 существенно выше, чем содержание Ba. Для LaLa1-линии (0.2666 нм) важно влияние CsLβ1- линии (0.2684 нм). Сложная ситуация имеет место для угловой позиции CeLa-линии, угловая позиция BaLβ1 (0.2568 нм) находится между CeLa1 (0.2562 нм) и CeLa2 (0.2571 нм) линиями. Для VKa1 - линии (0.2504 нм) необходимо учитывать наложение TiKb1-линии (0.2514 нм) для многих типов горных пород, хотя интенсивность TiKb1-линии примерно в 6 раз меньше интенсивности VKa1-линии. Причина состоит в неблагоприятном соотношении концентраций Ti и V (например, в СО ДВГ титана содержится в 48 раз больше, чем ванадия, в ДВД в 110 раз, в СГ-1 - в 144 раза, в СГ-3 - в 433.3 раза). Для LaLβ1-линии (0.2459 нм) могут возникать проблемы из-за близости PrLa1-линии (0.2463 нм). Линия BaLβ2 в качестве аналитической не представляет интереса, так как её интенсивность примерно в 4.5 раза меньше, чем для BaLa - и BaLβ1-линий. Перспективно использование в качестве аналитических линий CeLβ1 и NdLa (длины волн 0.2356 и 0.2370 нм, соответственно).
Из рисунка 2 видно, что для угловой позиции MoKa1 (0.0709 нм) существенно влияние наложения линии ZrKb1 (0.0702 нм). Для NbKa1-линии (0.0746 нм) важно рассмотреть влияние YKβ1-линии (0.074 Å). При определении содержания циркония (0.0786 нм) необходимо учесть наложение SrKb1 (0.0783 нм). При нахождении интенсивности YKa1 (0.0829 нм) следует учитывать влияние RbKb1 (0.08287 нм). В случае повышенного содержания (>0.05%) Zr возможно влияние хвоста ZrKa1-линии на аналитическую линию YKa1. Угловая позиция ULa1 (0.0911 нм) находится между RbKa1 (0.0926 нм) и SrKa1 (0.08753 нм) линиями. Содержание U в горных породах обычно существенно ниже содержаний Rb и Sr (17 ppm и 1500 ppm, 140 ppm - для СО ДВГ, 63 ppm и 1100 ppm, 18 ppm - для СО СГ-1А) и, кроме того, интенсивность Ka1-линии в три-четыре раза выше интенсивности La1-линии. Это приводит к необходимости учитывать влияние наложения RbKa1- и SrKa1- линий на ULa1-линию. ThLa1-линия (0.0956 нм) находится вблизи RbKa1 и при расчёте содержаний Th это также нужно учитывать. Для определения содержаний свинца в качестве аналитической линии выбрана PbLb1-линия (0.0983 нм). Хотя регистрируемая интенсивность PbLa1-линии несколько выше интенсивности PbLb1-линии, использование PbLa1-линии в качестве аналитической не возможно, так как длина волны PbLa1-линии (0.1175 нм) совпадает с длиной волны AsKa1-линии (0.1176 нм).
При определении содержания хлора и серы необходимо учитывать влияние L-линий Rh (Lα - 0.461 нм, Ll - 0.522 нм). С длинноволновой стороны от аналитической линии SКα1 (0,5347 нм) находится NbLβ1-линия (0.5492 нм). Влияние NbLβ1-линии на интенсивность аналитической линии SКα1 в данной методике не учитывалось, так как интенсивность Lβ1 линий в 3-5 раз меньше интенсивности Кα1 при условии близости длин волн этих линий. Кроме того, концентрация S в почвах и осадочных породах превышает концентрацию Nb в десятки раз (для СГХМ-1 содержания S и Nb соответственно составляют 400 и 12 ppm, для СДО-1 – 1200 и 10 ppm, для СП-1 – 300 и 15 ppm). С коротковолновой стороны от аналитической линии РКα1 (0.6157 нм) находится аналитическая линия ZrLα1 (0.6078 нм). Влияние интенсивности ZrLα1-линии на интенсивность РКα1 незначительно. При определении концентрации фосфора необходимо учитывать вклад в интенсивность РКα1 - линии излучения СаКβ1 - линии во втором порядке отражения (0.6179 нм). При разработке методики определения оксида калия учтено влияние RhLg2,3-линии (0.3686 нм) на интенсивность аналитической линии KKα1 (0.3744 нм).
Аналитическая линия GaKα1 относительно свободна от наложения других аналитических линий. При количественном определении Ga в пробах с повышенным содержанием цинка, необходимо учитывать влияние хвоста ZnKβ1,3-линии (0.1295 нм) на интенсивность аналитической линии GaKα1 (0.1344 нм). С длинноволновой стороны от аналитической линии GaКα1 находится CuKβ1,3 - линия (0.1393 нм). В данном случае линии хорошо разрешимы, интенсивность Кα - линии в 6-7 раз выше интенсивности Kβ1.
С коротковолновой стороны от аналитической линии CuKα1 (0.1541 нм) расположена Kβ1,3-линия никеля (0.1500 нм). В ультраосновных породах содержание никеля на несколько порядков выше содержания меди, поэтому необходимо учитывать влияние NiKβ1,3-линии на интенсивность аналитической линии меди.
Угловая позиция ZnKa1-линии (0.1435 нм) относительно свободна от наложения других аналитических линий. Влияние CuKb1-линии (0.1392 нм) на аналитическую линию Zn незначительно, так как, во-первых, соотношение интенсивностей линий Kα и Kβ равняется 7.5 : 1, во-вторых, для этих линий имеет место лишь частичное перекрывание.
Рентгеновский спектр в диапазоне длин волн от FeKb1 до CrKα1, полученный с использованием СО СГ-1, представлен на рисунке 3. Угловая позиция СoKα1 (0.1789 нм) находится между FeKb1 и FeKα1. Соотношения концентраций кобальта и породообразующего элемента Fe2O3(общее) составляет 1:1×103 – 1:4×103 (например в ДВМ – 0.012 и 12.55%, в СТи 15.22%, соответственно), поэтому необходимо учитывать влияние хвоста FeKb1-линии (0.1757 нм) на линию СoKα1. С коротковолновой стороны от угловой позиции MnKα1 (0.2102 нм) находится FeKα1-линия (0.1936 нм). Для количественного определения концентраций MnO важно учитывать влияние FeKα1- линии на аналитическую линию марганца. Для CrKα1-линии (0.2290 нм) в ряде случаев необходимо предусмотреть влияние наложения более слабой VKb1-линии (0.2284 нм).
При анализе спектров для каждого определяемого элемента выбраны аналитические линии и фоновые позиции, относительно свободные от наложения спектральных линий элементов, присутствующих в образце (табл. I). Из выше изложенного следует что, для точного расчета концентраций определяемых элементов необходимо учитывать вклады посторонних излучений в экспериментальную интенсивность аналитических линий (табл. II). При расчете градуировочных коэффициентов, учитывающих вклады мешающих линий, использовали интенсивности аналитических линий, выбранных для измерения интенсивностей определяемых элементов.
2.3. Приготовление излучателей
Анализируемый материал должен быть гомогенным по гранулометрическому и химическому составу (диаметр частиц около 50 мкм). Масса анализируемого материала, требуемого для анализа, составляет около 5 г. Анализируемый материал прессовался в таблетки одинаковой плотности. Использовался пресс HTP-40 фирмы HERZOG с нагрузкой 20 т. Для повышения прочности таблеток в пробу перед таблетированием добавлялось связующее вещество (воск) в соотношении 5:1. В качестве подложки для таблеток использовалась борная кислота.
2.4. Выбор параметров измерения аналитических линий определяемых элементов
Интервалы определяемых содержаний составляют (в %): оксид фосфора – 0.011-1.82, сера общая – 0.006-0.44, хлор – 0.004-1.7, оксид калия – 0.105-4.68, оксид титана – 0.0, ванадий – 0.00025-0.032, хром – 0.0005-0.2, оксид марганца – 0.014-1.125, кобальт – 0.0002-0.012, никель – 0.0006-0.а13, медь – 0.0004-0.31, цинк – 0.0024-0.2, галлий – 0.0005-0.004, рубидий 0.001-0.15, стронций – 0.0005-0.33, иттрий – 0.0005-0.015, цирконий – 0.002-0.063, ниобий – 0.0005-0.038, молибден – 0.001-0.026, барий – 0.0019-0.225, лантан – 0.0007-0.015, церий – 0.001-0.039, неодим – 0.001-0.007, свинец – 0.0005-0.013, торий – 0.0005-0.0130, уран – 0.0002-0.0063. Выбранные параметры измерений аналитических линий определяемых элементов представлены в таблице III. Использовали рентгеновскую трубку AG22 с Rh-анодом (толщина Be-окна 0.075 мкм). Мощность трубки была всего лишь 1 кВт. Это позволяло упростить систему охлаждения рентгеновской трубки. Оценка зависимости интенсивностей Ka - линий Ca, Ti, V, Fe, La - линий Ba, La, Nd и Lb - линии Ce и рентгеновского фона от потенциала на рентгеновской трубке выполнена для напряжений от 20 до 50 кВ. Выбранное время измерения интенсивностей аналитических линий определяемых элементов представлено в табл. 1. Хотя при определении концентраций P2O5, S, Cl, Cr, Co, Cu и Ga используются наиболее яркие Ka - линии характеристического спектра, выбранная экспозиция измерения их аналитических линий существенно превышает время измерения аналитических линий K, Ti, Mn. Это объясняется их различым содержанием в исследуемых горных породах. Количественное определение содержаний Ba, La, Ce, Nd, Th и U производится с использованием La - и Lb - линий и диапазон содержаний данных элементов в горных породах составляет от 0.0002 до 0.225 %. С учётом этих факторов время измерения, требуемое для регистрации излучения этих линий, необходимо было увеличить.
3. РАЗРАБОТКА МЕТОДИК ОПРЕДЕЛЕНИЯ КОНЦЕНТРАЦИЙ
3.1. Относительные удельные интенсивности
В работах [5,6] исследованы взаимные влияния элементов с использованием теоретических и экспериментальных интенсивностей для рассматриваемых типов горных пород. Полученные данные показали, что относительные удельные интенсивности для некоторых аналитических линий изменяются существенно в зависимости от типа горных пород. Первоначально в методике определения содержаний TiO2, MnO, V, Ba, La, Ce, Nd, Cu, Ga, Zn, Ni, P2O5, S, Cl и K2O для учёта взаимных влияний элементов предполагалось использовать интенсивности аналитических линий большинства из основных породообразующих элементов. Однако результаты теоретических оценок и экспериментальные данные показали, что снижение ошибок определения содержаний этих элементов при введении поправок на матричные эффекты с помощью интенсивностей аналитических линий Mg и Al получалось незначительным. Поэтому, для обеспечения приемлемых ошибок с помощью разрабатываемой методики для поправок на взаимные влияния элементов использовались только интенсивности Ka - линий Si, Ca и Fe.
При определении содержаний Mo, Nb, Zr, Y, Sr, Rb, U, Th и Pb для коррекции взаимных влияний элементов использовался способ стандарта фона [5]. При расчёте содержаний Nb, Zr, Sr в качестве стандарта использовалось некогерентно рассеянное на образце характеристическое излучение анода рентгеновской трубки (18.505°). Аналогично, в случае расчёта содержаний Y, Rb U, Th и Pb – интенсивность излучения, измеренная с длинноволновой стороны от угловых позиций определяемых элементов (2q=29°) [5-11].
3.2. Используемые калибровочные уравнения
Программное обеспечение рентгеновского спектрометра S4 EXPLORER позволяет выбирать оптимальные калибровочные уравнения с помощью программы Fquant и выполнять математическую обработку результатов измерения интенсивностей набора СО, используемого для калибровки методики. Интенсивности для этого набора измеряются с помощью программы Loader. В программном обеспечении FQuant для учёта матричных эффектов реализован способ, основанный на использовании - коэффициентов со следующим уравнением:
![]() , (1)
, (1)
где
Ÿ Ci и Ii - концентрация и интенсивность аналитической линии калибруемого элемента, исправленная на фон;
Ÿ mi – наклон калибровочного графика;
Ÿ Ij – интенсивности аналитических линий матричных элементов, исправленные на фон;
Ÿ - альфа-коэффициенты, используемые для учёта матричных эффектов и вычисленные с помощью уравнения линейной регрессии.
При калибровке уравнений с помощью метода наименьших квадратов в качестве параметра минимизации использованы относительные квадратичные отклонения концентраций:
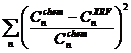 , (2)
, (2)
где
· ![]() - аттестованные значения концентрации n-го СО;
- аттестованные значения концентрации n-го СО;
· ![]() - вычисленные значения концентрации для этого СО.
- вычисленные значения концентрации для этого СО.
3.3. Выбор стандартных образцов для калибровки
Стандартные образцы, используемые для калибровки определяемых элементов, выбирались из коллекции СО кислого, основного, ультраосновного, карбонатно-силикатного и карбонатного составов, имеющихся в наличии в ЦАЛ БГРЭ. При расчёте градуировочных характеристик для каждого определяемого элемента некоторые СО были исключены из калибровочного массива по следующим причинам:
1. Значение концентрации определяемого элемента в данном СО не аттестовано или аттестованное значение концентрации определяемого элемента ниже предела обнаружения.
2. Содержание определяемого элемента определено ориентировочно, т. е. число результатов измерений для этого образца было недостаточным на стадии аттестации.
3. Матрица СО резко отличается от матриц образцов основного калибровочного массива.
В калибровке использовались данные по химическому составу стандартных образцов
из работ , [12] и Govindaraju K. [13]. В таблице II представлена информация о градуировочных характеристиках определяемых элементов.
Коэффициенты для учёта вклада наложения линий, длины волн которых близки к длинам волн определяемых элементов, вычисляли по программе Fquant с помощью уравнения:
![]() , (3)
, (3)
где
· ![]() - интенсивность аналитической линии определяемого элемента, исправленная на фон;
- интенсивность аналитической линии определяемого элемента, исправленная на фон;
· ![]() - интенсивность налагающейся линии, исправленная на фон (вычисленная или измеренная);
- интенсивность налагающейся линии, исправленная на фон (вычисленная или измеренная);
· - коэффициент регрессии.
Предусмотрена возможность использования смешанного варианта коррекции, состоящего из нескольких имеющихся в программном обеспечении типов коррекции.
Представлены графики зависимости между интенсивностями аналитических линий и концентрациями для серы и урана (рис. 4, 5). Сравнивая градуировочные зависимости, построенные без учёта взаимных влияний элементов (рис. 4а, 5а) и градуировочные графики после введения поправок на взаимные влияния элементов (рис. 4b, 5b), можно видеть существенное уменьшение разброса точек на графиках после введения поправок.
4. РЕЗУЛЬТАТЫ
Для оценки точности результатов анализа во всем диапазоне содержаний определяемых элементов проведены метрологические исследования [14, 15]. При оценке точности результатов анализа оценивались следующие метрологические характеристики методики:
· Суммарное среднее квадратическое отклонение относительной погрешности ![]() ;
;
· Среднее квадратическое отклонение относительной случайной составляющей погрешности ![]() ;
;
· Среднее квадратическое отклонение части относительной случайной составляющей погрешности![]() ;
;
· Среднее квадратическое отклонение относительной не исключенной систематической составляющей погрешности ![]() ;
;
· Систематическая составляющая относительной погрешности, выраженная средним арифметическим расхождением результатов анализа от установленного содержания компонента в пробе, ![]() (систематическая погрешность).
(систематическая погрешность).
Метрологические исследования методик количественного определения содержаний исследуемых элементов проводились методом дисперсионного анализа с использованием набора СО и аттестованных смесей (АС).
В одну выборку объединялись результаты, соответствующие нескольким табличным диапазонам, при этом проверялось статистическое равенство (или неравенство) дисперсий при уровне значимости a = 0,05 и доверительной вероятности Р = 95%. При объединении нескольких табличных диапазонов в один общий диапазон на каждый табличный диапазон содержаний приходилось не менее двух СО и АС (k ![]() 2). Однородности дисперсий оценивались по критерию Кохрена.
2). Однородности дисперсий оценивались по критерию Кохрена.
Выборочные дисперсии считаются однородными и экспериментальные данные объединяются в одну совокупность, если выполняется неравенство Gmax![]() Gтабл., где Gmax вычислялось по формуле:
Gтабл., где Gmax вычислялось по формуле:
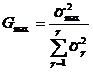 , (4)
, (4)
где ![]() - выборочная дисперсия для каждой пробы,
- выборочная дисперсия для каждой пробы, ![]() - максимальная из вычисленных дисперсий, Gтабл – табличное значение для уровня значимости a = 0,05.
- максимальная из вычисленных дисперсий, Gтабл – табличное значение для уровня значимости a = 0,05.
Результаты определения для каждой совокупности обрабатывались методом двухфакторного дисперсионного анализа. Значимость систематической составляющей погрешности x оценивалась для каждого табличного диапазона содержаний по критерию ”ничтожной погрешности”.
В большинстве случаев выборочная дисперсия, характеризующая повторяемость определений, обусловлена неконтролируемыми факторами, медленно изменяющимися во времени. Величина среднеквадратического отклонения относительной погрешности в большей степени зависит от вариаций общего химического состава образцов, так как численные величины выборочной дисперсии, характеризующей повторяемость определений, значительно меньше численных величин выборочной дисперсии, характеризующей избирательность методики.
Чувствительность методики характеризуется пределом обнаружения, который рассчитывался по формуле:
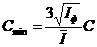 , (5)
, (5)
где ![]() - разность между средним значением интенсивности, измеренным на месте аналитической линии, и интенсивностью фона
- разность между средним значением интенсивности, измеренным на месте аналитической линии, и интенсивностью фона ![]() ; C – концентрация определяемого элемента. Для выбранных условий (существенно, что используемая мощность рентгеновской трубки всего лишь 1 кВт) предел обнаружения, рассчитанный по критерию 3s, составил (в ppm): 0.5 для Zr, 0.6 для Y, 0.7 для Sr, 0.8 для Rb, 1 для V и Co, 1.3 для Th, 1.4 для Ni, Zn, Nb и U, 1.5 для S(общ.), Ga и Mo, 1.8 для Pb, 2 для Cr и Cu, 2.5 для MnO, 3 для Cl, 6 для TiO2 и P2O5, 12 для Ba, 5 для La и K2O, 7 для Ce и 9 для Nd. Эти данные близки к данным, полученным с использованием мощных рентгеновских трубок.
; C – концентрация определяемого элемента. Для выбранных условий (существенно, что используемая мощность рентгеновской трубки всего лишь 1 кВт) предел обнаружения, рассчитанный по критерию 3s, составил (в ppm): 0.5 для Zr, 0.6 для Y, 0.7 для Sr, 0.8 для Rb, 1 для V и Co, 1.3 для Th, 1.4 для Ni, Zn, Nb и U, 1.5 для S(общ.), Ga и Mo, 1.8 для Pb, 2 для Cr и Cu, 2.5 для MnO, 3 для Cl, 6 для TiO2 и P2O5, 12 для Ba, 5 для La и K2O, 7 для Ce и 9 для Nd. Эти данные близки к данным, полученным с использованием мощных рентгеновских трубок.
По результатам метрологических исследований можно сделать следующие выводы.
1. Для определения концентраций TiO2, V, Ba, La, Ce, Nd, Co, MnO, Cr, Ni, Zn, Mo, Nb, Zr, Y, Sr, Rb, U, Th и Pb в следующих диапазонах содержаний (в ppm): 100-190 и , 100-990, 20-1900, 5-190, 10-490, 20-49, 2-190, , 5-19 и 100-990, 5-190, 20-1900, 10-490, 5-490, 20-990, 5-190, 10-4900, 10-1900, 20-99, 5-99, 5-490, соответственно, данные методики могут быть аттестованы в качестве методик выполнения измерений I категории точности.
2. Для определения концентраций TiO2, V, Nd, MnO, Cr, Ni, U, Th, Ga и Cu, в диапазонах содержаний (в ppm) , 5.6-99, 50-99, , 20-99 и , , 2-19, 100-190, 5-49, 10-190, соответственно, данные методики могут быть аттестованы в качестве методик выполнения измерений II категории точности.
3. Для методик определения Ga, Cu, K2O, P2O5, S и Cl проведены предварительные метрологические исследования. Полученные результаты для Ga в диапазоне от 5 до 49 ppm и Cu в диапазоне от 10 до 190 ppm соответствуют II категории точности, для K2O в диапазоне концентраций ppm - III категории точности. Необходимо продолжить расчёт метрологических характеристик отдельных элементов для других диапазонов содержаний.
Ранее авторы данной работы докладывали о разработанных методиках количественного определения содержаний некоторых из рассматриваемых в настоящем сообщении элементов, в частности: TiO2, V, Ni, Zn, Rb, Sr, Y, Zr, Nb, Mo, Ba, La, Ce, Nd, Pb, Th, U [10, 11, 16, 17].
5. ЗАКЛЮЧЕНИЕ
Разработанные методики внедрены в аналитическую практику Центральной аналитической лаборатории Ботуобинской геологоразведочной экспедиции АК «АЛРОСА». Пределы обнаружения для определяемых элементов оказались близкими по величине данным, полученным с использованием мощных рентгеновских трубок.
Литература
1. Доусон Дж. Кимберлиты и ксенолиты в них. М.: Мир, 19с.
2. Сборник инструкций “SPECTRAPLUS” для пользователей спектрометра S4 EXPLORER.
3. , Пожиганова лабораторияТ. 69, № 6. - С. 70-72.
4. , Кремер лабораторияТ. 70, № 6. - С. 21-22.
5. Ревенко флуоресцентный анализ природных материалов. Новосибирск: ВО Наука. Сиб. издательская фирма. 19с.
6. , , // Аналитика и контроль. – 2001. № 5. - С. 409-416.
7. , , Жалсараев определение Rb, Sr, Y, Zr, Nb, Sn, Ba, La, Ce в горных породах на энергодисперсионном спектрометре с поляризатором // Аналитика и контроль, 6 (4). - С. 400-407.
8. , , Ревенко определение некоторых следовых элементов в горных породах на энергодисперсионном спектрометре с поляризатором // Proceedings of the ISCP-2, Ulaanbaatar. -2002. - С. 132-145.
9. , , Сопоставление результатов измерений Zr, Nb, Y, Rb, Ba, Ni в базальтах и трахитах методами ICP-MS и рентгеновской флуоресценции. Земная кора - 1996. - ИркутскС. 81-83.
10. Revenko A. G., Hudonogova E. V., Cherkashina T. Yu., Budaev D. A. X-ray fluorescence determination of Rb, Sr, Y, Zr, Nb, Mo, Pb, Th, and U in various types of rocks, Proceedings of SPIE-II. Zvenigorod, RussiaP. 7.
11. , , Будаев определение Ti, V, Ba, La, Ce, Nd, Nb, Zr, Y, Sr, Rb, Th, U и Pb в различных типах горных пород. VII Конференция “Аналитика Сибири и Дальнего Востока”. Новосибирск, Т. 2. - С. 61.
12. , Петров образцы состава природных сред. Новосибирск: Наука, Сиб. отделение, 19с.
13. Govindaraju K. Geostandards Newsletter: Special Issue, 1994; 18, 158 P.
14. ОСТ 9 «Методики количественного химического анализа».
15. ГОСТ Р ИСО 5725-1 – 2002 «Точность (правильность и прецезионность) методов и результатов измерений. Часть 1. Основные положения и определения».
16. Revenko A. G., Hudonogova E. V., Cherkashina T. Yu., Budaev D. A. X-ray fluorescence determination of Ti, V, Ni, Zn, Ba, La, Ce and Nd in various types rocks // In “Book of abstracts: European Conf. on X-Ray Spectrometry. Alghero, Italy. 2004. P. 204.
17. , , Будаев определение Ti, V, Ni, Zn, Rb, Ba, La, Ce, Nd, Pb в различных типах горных пород // В кн.: Тез. Докл. Всерос. Конф. по аналитической химии «Аналитика России 2004». М. 2004. С. 263-26.
