
Партнерка на США и Канаду по недвижимости, выплаты в крипто
- 30% recurring commission
- Выплаты в USDT
- Вывод каждую неделю
- Комиссия до 5 лет за каждого referral
Новые металлсодержащие нанокомпозиты: сенсорные и диэлектрические свойства
1, 2, 1, 1, 2, 1, 1,2, Yu. Feldman3
1 Научно-исследовательский физико-химический институт им. , Москва, , *****@***nifhi. *****
2 Институт химической физики им. РАН, Москва, ул. Косыгина, 4,
3 Hebrew University, Jerusalem, 91904, Israel
Изучены свойства сенсоров на основе полупроводниковых оксидных пленок, полученных методом аэрозольного напыления. Проводимость в воздухе пленок с наночастицами оксидов различных металлов, такими как Sn, In, Pd и др., селективно и резко изменяется при добавлении даже очень малых количеств различных химических соединений (Н2, CO, СН4, NH3, C2H5OH, H20, О3 и т. д.). Этот эффект обусловлен изменением механизма процессов переноса заряда между наночастицами, связанных с адсорбцией газов на поверхности наночастиц металла, присутствующих в композите. В частности, изучен сенсорный отклик пленки диоксида олова, содержащей нанокристаллы Pd, на различные концентрации водорода. Сопротивление пленки уменьшается под влиянием водорода, что обусловлено диссоциативной адсорбцией молекул водорода на нанокристаллах Pd и изменением работы выхода электрона из этих нанокристаллов. Проведенные исследования позволили разработать новый метод получения высокоэффективных сенсоров для детектирования газов-восстановителей, обладающих исключительно высокой скоростью отклика. Так, время отклика сенсоров на водород при различных концентрациях не превышает 1 сек.
Развиты новые теоретические подходы для описания диэлектрических свойств композиционных материалов на основе диэлектрической матрицы, содержащей наночастицы металла и/или полупроводника. В соответствии с этой теорией, в равновесном состоянии электрон переходит из нанокластеров в ловушки, расположенные внутри диэлектрической матрицы вблизи кластера. Электрон и кластер, связанные между собой кулоновскими и поляризационными силами, образуют элементарный диполь. Число и величина таких равновесных диполей зависят от температуры. Решение интегрального уравнения, выведенного для временной функции релаксации диполей, зависит от диэлектрических характеристик матрицы и распределения наночастиц по размерам, которые могут быть определены экспериментально. Характеристическое время туннелирования электрона между соседними ловушками также является функцией вышеуказанных параметров. Неизвестными величинами являются энергия связи электрона в ловушке и плотность ловушек в матрице. Развитая теория использована для интерпретации экспериментальных данных, полученных при исследовании диэлектрических свойств пористых неорганических силикатных стекол с контролируемым размером пор, содержащих 4-6 масс.% наночастиц палладия. Распределение пор по размерам, а также состав исследованных образцов определяли методами сканирующей электронной и туннельной микроскопии. Экспериментальные данные, полученные методом диэлектрической спектроскопии в температурном интервале -120 – 300оС и частотном интервале 0,1 – 105 Гц, хорошо согласуются с теоретическими закономерностями комплексной диэлектрической проницаемости.
Работа поддержана РФФИ (гранты ; -офи; ).
Мономолекулярные реакции в плотных средах.
М.
Институт проблем химической физики РАН, Черноголовка.
Рассмотрены механизмы влияния плотного молекулярного окружения в сжатых высоким давлением газах и жидкостях, в твердых веществах и полимерах на скорость мономолекулярной реакции.
Традиционно эта проблема рассматривается в рамках теории Эванса-Поляни [1], впервые предложенной Вант Гоффом [2]. Предполагается, что при образовании АК происходит деформация окружающей среды и дополнительная энергия, рассчитанная методами равновесной термодинамики, добавляется к энергии активации реакции. Но время существования АК »10-13 с, а характерные времена молекулярных движений порядка 10-10 – 10-8 с, и потому применять равновесную термодинамику к процессу образования АК в плотных молекулярных средах нельзя.
Предлагается иная модель. Реакция протекает на фоне неподвижного молекулярного окружения. Если реакция протекает с увеличением объема, т. е. реакция имеет положительный объем активации, то реакция происходит, если вблизи молекулы есть свободный пространство, достаточное для образования АК, если такого пространства нет, то реакция не происходит. Таким образом, проблема влияния молекулярного окружения на скорость мономолекулярной реакции с положительным объемом активации сводится к определению вероятности образования полостей необходимого размера. В данной работе она оценивалась в рамках модели «свободного объема» [3].
Показано, что традиционная теория справедлива для газов. В жидкостях объемы активации, вычисленные по предлагаемой теории, в 4 – 6 раз выше, чем объемы активации, рассчитанные по теории Эйринга.
В реакциях, идущих с уменьшением объема, то есть с отрицательным объемом активации, влияние давления осуществляется путем упругого деформирования молекулы по координате реакции. Это приводит к квадратичной зависимости lnk от давления.
В гетеролитических процессах добавляются два дополнительных механизма: электрострикция и поляризация среды, снижающих энергию заряженных систем по сравнению с энергией этих систем в вакууме, причем в процессе образования АК участвует только электронная поляризация, а в образовании заряженных долгоживущих состояний – продуктов или реагентов, дополнительно участвуют электрострикция и ориентационная поляризация. Это приводит к тому, что в гетеролитических процессах объемы активации прямой и обратной реакции не равны по абсолютной величине, процессы с образованием ионов из нейтральных молекул имеют большие объемы активации, чем обратные процессы. Поэтому высокое давление ускоряет гетеролитические процессы, идущие с образованием ионов.
Литература.
1. Evans M. G., Polanyi M. – Trans. Farad. Soc., vol. 31, 1935, p. 875-881.
2. Vant’ Hoff J. H. Lectures on Theoretical and Physical Chemistry. Braunschweig, 1901.
3. Теория абсолютных скоростей реакций, М.; ИЛ, 1948.
ТЕОРЕТИЧЕСКОЕ МОДЕЛИРОВАНИЕ МЕХАНИЗМА ИЗОМЕРИЗАЦИИ КАТИОН-РАДИКАЛОВ О - И М-ЭТИЛТОЛУОЛОВ
, ,
Казанский государственный технологический университет,
Казань, ул. К. Марса 68, *****@***ru
Были изучены реакции, относящиеся к первичным стадиям механизма распада катион-радикалов этилтолуолов. Расчеты проводились методом B3LYP/6-31G(d) с помощью пакета прикладных программ Gaussian98 [1]. Для катион-радикалов о - и м-этилтолуолов величина энтальпии активации мономолекулярного переноса водорода от этильного заместителя выгоднее, чем от метильного заместителя в ароматическом кольце. Цифры над стрелками и под соединениями – относительные энтальпии образования переходных состояний реакций и соединений (в кДж/моль). За ноль принята энтальпия образования о - этилтолуола.
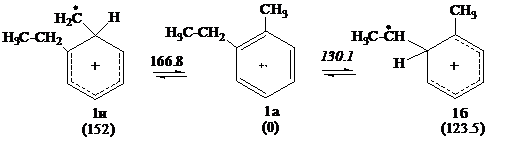
Нами были найдены переходные состояния для реакций изомеризации в катион-радикалах о - и м-этилтолуолов, которые доказывались присутствием одного отрицательного значения в матрице Гессе и подтверждались спусками по координате реакции к исходным и конечным продуктам.

1. Frisch M. J., Trucks G. W. et al.: Gaussian 98 (Revision A.7). Gaussian Inc. Pittsburgh PA. 1998.
Безызлучательная дезактивация низших синглетных pp*-электронных состояний и эффект тяжелого атома
, , ,
, ,
Федеральное государственное унитарное предприятие
«Ордена Трудового Красного Знамени научно-исследовательский
физико-химический институт имени »,
Россия. Москва. 105064. Воронцово поле 10.
E-mail: *****@***ru
Люминесцентные свойства соединений существенно зависят от безызлучательной дезактивации электронных состояний. Так, напрмер, безызлучательная S-T (синглет-триплетная) конверсия, S1~~>T1, обычно приводит к гашению флуоресценции и возгоранию фосфоресценции. Согласно экспериментальным спектроскопическим и магнито-оптическим исследованиям введение в молекулу тяжелых (многоэлектронных) атомов приводит обычно к увеличения константы скорости KST конверсии (эффект тяжелого атома). Из скоростей (KsST) заселения триплетных подуровней (s = x, y, z) низшего триплетного состояния Ts, скорость (KzST), которая соответствует неплоскому спиновому состоянию, оказывается на порядок меньшей, чем другие. Результаты экспериментальных исследований, выявляющие эффект тяжелого атома для переходов S(pp*)~~>T(pp*), объясняют влиянием вибронных взаимодействий (VIB) на скорость KST конверсии.
Безызлучательная S-T конверсия описывается с привлечением «золотого правила Ферми», и для теоретических расчетов KST в настоящее время в литературе разработана модель чисто спин-орбитальных (SO) взаимодействий (основная модель) и модель вибронно-индуцированных спин-орбитальных (VISO) взаимодействий. Однако, особенность основной модели состоит в том, что для переходов S(pp*)~~>T(pp*) она не можеть передать эффект тяжелого атома. Кроме того, такая модель (согласно правилам отбора) позволяет рассчтывать только константы KzST. Широкими возможностями обладает модель VISO взаимодействий, однако использование этой модели отвергают, т. к. из-за грубых приближений в расчете VIB взаимодействий получают очень малые величины KsST. В результате, результаты экспериментальных исследований не находят теоретического описания.
В нашей работе усовершенствованы расчеты в рамках модели VISO взаимодействий, что позволило получать теоретические значения KST и KsST, согласующиеся с экспериментальными, т. е. описать эффект тяжелого атома, а также разную скорость заселенности всех трех триплетных подуровней Ts. Результат получен при учете полного VIB взаимодействия: учете для каждой молекулы всех неплоских колебательных мод с их сложной формой (вместо обычного использования двухатомных осцилляторов групп C-H). Кроме этого, вместо теории возмущений, нами использован прямой метод расчета VIB взаимодействий, который использовался в литературе для описания излучательных переходов. Расчеты выполнены на примере молекул дибензо-п-диоксина, дибензофурана и их некоторых хлопроизводных, а также на примере аценов (антрацен, нафталин) и из хлорзамещенных.
Работа выполнена при финансовой поддержке Российского фонда фундаментальных исследований (проект )
ТЕХНОЛОГИЯ ПРЕДСКАЗАНИЯ ПРОСТРАНСТВЕННОЙ СТРУКТУРЫ И ФИЗИКО-ХИМИЧЕСКИХ СВОЙСТВ МОЛЕКУЛЯРНЫХ ВЕЩЕСТВ В ТВЕРДОМ СОСТОЯНИИ
Физико-химический институт им. ,
Москва, , E-mail: *****@***nifhi. *****
В основе физико-химических свойств вещества, проявляемых в твердом состоянии, лежит его кристаллическая структура. Однако рентгеноструктурные исследования кристаллических веществ с атомным разрешением в большинстве своем ограничены редкими в общей массе случаями получения образцов в виде совершенных монокристаллов. Еще меньше возможности экспериментальных методов в получении структурной информации в условиях экстремальных давлений и температур. Поэтому прогресс в индустрии наносистем и материалов невозможен без опережающего развития методов численного моделирования структур на атомно-молекулярном уровне, где эти методы призваны играть роль инструментов теоретической проработки и конструирования новых наносистем, аналогично тому, как в строительной индустрии сооружению зданий из строительных материалов предшествует этап проектирования в архитектурной мастерской или конструкторском бюро.
В докладе дан обзор текущего состояния методов предсказания кристаллической структуры и свойств молекулярных веществ расчетным путем на основе глобальной минимизации свободной энергии, вычисляемой в попарно-аддитивном приближении с полуэмпирическими атом-атомными потенциалами. Основная часть доклада посвящена работе, проделанной в Карповском институте, начало которой было положено еще в середине 1970-х годов. За прошедшее время разработанный нами метод и программы для ЭВМ показали свою высокую эффективность, что подтверждается примерами успешных предсказаний структур кристаллов, в числе которых продукты твердофазной полимеризации фуллеренов C60 и C70.
В настоящее время это направление получило новый импульс развития, что связано с появлением новых поколений вычислительной техники (суперкомпьютеры на базе кластеров ВМ, распределенные сети и т. п.), позволяющие увеличить скорость вычислений на порядки. Это открывает перспективы для решения задачи предсказания реальной структуры твердых состояний веществ, содержащих точечные и размерные дефекты упаковки, микрокристаллические домены, аморфную фазу и т. п.
В инженерных расчетах упругих, прочностных, тепловых и прочих характеристик механизмов, инженерных и строительных конструкций, сооружаемых из новых материалов, остро стоит проблема дефицита термодинамических и физических констант новых материалов (модули упругости, коэффициенты теплопроводности и теплового расширения и т. п.), а также их зависимости от параметров внешней среды (уравнения состояния). Получение этих констант экспериментальным путем сопряжено с большими затратами, особенно если речь идет об экстремальных условиях внешней среды. Применение методов моделирования атомно-молекулярной структуры и динамики кристаллических твердых веществ обещает совершить настоящий прорыв в решении данной технически важной проблемы.
Литература
От молекулы к твердому телу: предсказание структур органических кристаллов Ж. физической химии 2008, Т. 82, No. 10, С. 1861–1870.
Фотодиссоциация малых ароматических молекул: роль внутренней конверсии в общей цепи фотохимических превращений
, Y.T. Lee, S.H. Lin, C.K. Ni
Institute of Atomic and Molecular Sciences, Academia Sinica, Taipei, Taiwan
e-mail: *****@***ru
В данной работе представлены результаты исследования механизмов фотоизомеризации и фотодиссоциации простейших ароматических молекул в молекулярных пучках (в отсутствие столкновений между молекулами) при их облучении высокоэнергетичными УФ-фотонами. Исследованы три серии химических соединений:
- бензол (C6H6) и его азот-замещенные производные: пиридин (NC5H5), пиримидин (N2C4H4) и триазин (N3C3H3);
- ароматические соединения, содержащие NH-группу в составе пятичленного кольца: индол (C8H7N), 1H-индол-3-этанамин (C10H12N2) и триптофан (C11H12N2O2);
- ароматические соединения, имеющие OH-группы: фенол (C6H5OH), нафтол (C10H7OH) и 2,5-дигидроксиацетофенон (C8H8O3).
Было установлено, что после возбуждения всех трех типов молекул УФ-фотонами на один из возбужденных электронных уровней возможен их безызлучательный переход в основное состояние посредством последовательной внутренней конверсии. Внутренняя конверсия становится возможной благодаря сильному сближению электронных термов основного и возбужденного уровней при определенном изменении геометрии молекул.
Для первой группы соединений таким изменением геометрии является раскрытие ароматического кольца. В частности, в такой конфигурации имеет место сильное сближение основного и первого возбужденного термов, в результате которого молекулы, находящиеся в первом возбужденном состоянии, могут релаксировать в основное состояние с образованием с равной вероятностью либо открытой бирадикальной структуры, либо исходной конфигурации с закрытым кольцом. Несмотря на незначительную величину обратного барьера перехода бирадикальной структуры в основную конфигурацию с закрытым кольцом, конечный выход продуктов диссоциации зависит от того, в какой из двух изомеров релаксировала молекула после внутренней конверсии. Различными также являются функции распределения кинетической энергии продуктов реакции.
Для второго типа соединений сближение основного и первого возбужденного термов происходит при увеличении длины связи N – H. При этом в точке сближения ППЭ первого возбужденного уровня имеет слабый минимум, а ППЭ основного состояния имеет максимум. После внутренней конверсии в основное состояние, молекула с равной вероятностью либо возвращается в исходную геометрию, либо теряет атом водорода. Этот процесс легко определяется в масс-спектрометрических экспериментах по наличию быстрой и медленной компонент в линии, соответствующей отрыву атома водорода.
Для третьей группы соединений сближение термов наблюдается при увеличении длины связи O – H. Процесс диссоциации сходен с процессом диссоциации второй группы соединений.
МАТЕМАТИЧЕСКАЯ МОДЕЛЬ ИСТЕЧЕНИЯ ЖИДКОСТИ ИЗ ЦИЛИНДРА
, Аунг Зо Мо,
«МАТИ» - Российский государственный технологический университет им.
г. Москва, ул. Оршанская, 3, МАТИ
Рассмотрена обобщенная математическая модель процесса истечения жидкости из вертикально расположенного цилиндрического резервуара высоты H, переменного сечения S(h) через систему регулируемых и нерегулируемых отверстий на дне и боковой поверхности.
Истечение, происходящее через расположенное на дне единственное регулируемое отверстие площади s(h) со скоростью V(t), описывается задачей Коши для обыкновенного дифференциального уравнения
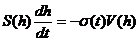 , (1)
, (1)
где h(t) – уровень жидкости в момент времени t. Наличие нескольких разноуровневых отверстий предполагает решение системы уравнений вида (1). Считается, что истечение жидкости подчиняется закону Торричелли
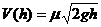 , (2)
, (2)
где m - коэффициент расхода жидкости (0 < m < 1).
Зависимость гидродинамических характеристик процесса истечения жидкости из цилиндра для широкого класса изменяющихся параметров исследована численно и аналитически.
Для гармонического и кусочно-линейных законов истечения жидкости из цилиндра через регулируемое отверстие найдены размеры эквивалентных цилиндров с единственным нерегулируемым отверстием на дне.
Установлены принципы нелинейной декомпозиции гидродинамических состояний системы для сложных законов истечения.
Найденные законы истечения жидкости из резервуаров могут быть использованы при исследовании широкого класса задач физической химии.
Литература
1. Нигматулин Рс. И., Соловьев гидромеханика: Учебное пособие. – М.: ГЕОТАР, 2005. – 512 с.
2. Зотов операций в прикладной гидродинамике/ В кн.: V Московская международная конференция по исследованию операций: Труды. – М.: МАКС Пресс, 2007, с. 1
3. Зотов декомпозиция процесса истечения жидкости из резервуара – Обозрение прикл. и промышл. матем., 2007, т. 14, в. 3, с. 533 – 534.
4. Зотов жидкости из резервуара через регулируемое отверстие / В кн.: Актуальные задачи математического моделирования и информационных технологий. – Сочи, 2008, с. 133 – 135.
Проводимость нанокомпозитов с ферромагнитными кластерами
1, 1,2
1Институт химической физики им. РАН, 119991 Москва, , *****@***com
1Научно-исследовательский физико-химический институт им. , 105064 Москва, , *****@***nifhi. *****
Развита теория проводимости композитов изолятора с нанокластерами ферромагнитных металлов, получена зависимость удельного сопротивления от температуры, электрического и магнитного полей. Проводимость осуществляется за счёт туннельных перескоков электронов между кластерами. Поскольку плотность состояний электронов в кластерах зависит от направления спина электрона по отношению к направлению намагниченности кластера, константа скорости перескока зависит от направления спина, а ток – от величины магнитного поля, выстраивающего намагниченности отдельных кластеров. Таким образом, сила тока в таких системах является заметной функцией магнитного поля, что приводит к возникновению гигантского магнитного сопротивления (ГМС).
У достаточно больших металлических нанокластеров, когда радиус частицы ![]() ≥ 1 нм, а именно такие кластеры рассматриваются в данной работе, уровень Ферми EF и магнитный момент, приходящийся на один атом ферромагнитного кластера, не зависят от размера кластера. Следовательно, скачки добавочного электрона по нейтральным кластерам не требуют теплового возбуждения. Однако, для того чтобы оторвать электрон от нейтрального кластера и переместить его на другой кластер, требуется затратить энергию ε+
≥ 1 нм, а именно такие кластеры рассматриваются в данной работе, уровень Ферми EF и магнитный момент, приходящийся на один атом ферромагнитного кластера, не зависят от размера кластера. Следовательно, скачки добавочного электрона по нейтральным кластерам не требуют теплового возбуждения. Однако, для того чтобы оторвать электрон от нейтрального кластера и переместить его на другой кластер, требуется затратить энергию ε+ ![]() е2/Rcl, порядка нескольких десятых эВ, т. е. много больше kT.
е2/Rcl, порядка нескольких десятых эВ, т. е. много больше kT.
Можно выделить три чётко разделённых режима прохождения тока через систему кластеров: 1) режим слабого электрического поля, eUif << kT , где Uif – средняя разность потенциалов между соседними кластерами; 2) режим сильного электрического поля, ε+ > eUif >> kT; 3) режим сверхсильного электрического поля, Uif >ε+. В первом режиме ток пропорционален равновесной концентрации добавочных электронов на нейтральных кластерах и прыжковой подвижности электронов, а элементарное сопротивление определяется обратной вероятностью прыжка. Во втором режиме электрон может прыгать по нейтральным кластерам только в сторону анода, и элементарное сопротивление – это сопротивление цепочки, начинающейся с ионизации кластера и серии скачков рождённого электрона по нейтральным кластерам. Третий режим аналогичен второму, но с более короткой цепочкой. Рассчитана зависимость удельного сопротивления от T и Uif для всех режимов.
Поскольку сопротивление цепочки кластеров складывается из отдельных элементарных сопротивлений, то и зависимость от магнитного поля следует искать именно для среднего элементарного сопротивления, а не для средней проводимости, как это делалось ранее и в результате получалось завышенное значение ГМС. Выведено выражение для ГМС плёнки композита. Численные расчёты показывают, что, например, наложение магнитного поля напряженностью 1,25 кГс при комнатной температуре на композиты, содержащие кластеры железа радиусом 3,3 нм, приводит к уменьшению сопротивления на 23 %.
Работа поддержана РФФИ (гранты ; -офи; ).
ФЛИККЕР-ШУМОВАЯ СПЕКТРОСКОПИЯ В АНАЛИЗЕ ПРИРОДЫ СОЛНЕЧНОЙ АКТИВНОСТИ
Г., ,
Научно-исследовательский физико-химический институт им. Л.Я. Карпова,
105064 Москва, ул. Воронцово поле 10. *****@
Одним из достоинств метода фликкер-шумовой спектроскопии (ФШС) является возможность разделять вклад в вариации наблюдаемой переменной резонансной (периодической) и хаотической составляющих. Такая возможность появляется благодаря разработанной идее совместного анализа свойств спектра мощности временного ряда измеряемой переменной и его структурной функции (разностного момента второго порядка). Это особенно важно, когда анализируются временные ряды такой переменной, для которой хаотическая составляющая вариаций имеет существенно меньшую амплитуду, чем периодическая.
Примером таких рядов являются ряды наблюдаемых вариаций различных параметров солнечной активности. Для них, как широко известно, наиболее ярко выраженной является квазипериодическая 11-летняя вариация, амплитуда, период и форма которой изменяются от цикла к циклу, и природа которой остается непонятой. Существующая теория турбулентного магнитного динамо в принципе объясняет возможность периодических вариаций наблюдаемых параметров солнечной активности, однако в рамках этой теории не удается получить наблюдаемые период и вариабельность свойств цикла. Исследование свойств хаотической динамики солнечной активности на разных временных шкалах может дать новую информацию для понимания ее источников. В работе проанализированы самые длинные (250лет наблюдений) из накопленных рядов вариаций солнечной активности - ряд ежедневных и среднемесячных значений чисел Вольфа, характеризующие динамику выхода на солнечную поверхность сильных магнитных полей. В качестве феноменологических параметров при параметризации хаотического сигнала использовались: n -спектральный индекс (показатель наклона спектра мощности) и H1 - константа Хёрста, определяемая из структурной функции. Значения n и H1 были получены для нескольких частотных интервалов, причем в низкочастотном интервале, где вклад периодической составляющей существенен, применена вычислительная процедура, разработанная в ФШС. Показано, что:
- в вариациях значения чисел Вольфа хаотическая динамика проявляется на всех временных шкалах, причем спектральный индекс является параметром, чувствительным к изменению ее характера;
- для рассмотренного процесса в частотном интервале 0.5 год-1<f~< 10 год-1 (f-частота) не выполняется общепринятое соотношение 2H1 = n-1, что связано с преобладающим вкладом в структурную функцию крупномасштабных хаотических флуктуаций измеряемой динамической переменной;
-значение спектрального индекса n изменяется на временах порядка 2 года, что совпадает с характерным временем продолжительности солнечного цикла, получаемого в теории солнечного динамо. Этот результат позволяет предположить, что существует процесс, который медленно изменяет условия функционирования солнечного динамо, т. е. указывает на существование нестационарных условий магнитной конвекции на Солнце на временных масштабах больше нескольких лет;
- делается предположение о природе такой не стационарности.
ПОЛИМЕРНЫЕ МОЛЕКУЛЯРНЫЕ КОМПОЗИТЫ – НАДЕЖДЫ И РЕЗУЛЬТАТЫ
,
Институт нефтехимического синтеза РАН
119991 Москва, Ленинский проспект, 29
email: *****@
Крупнейшим достижением отечественной химической науки и технологии в прошлом веке было создание и организация промышленного производства высокопрочных волокон СВМ и Армос на основе жесткоцепного полимера - полиамидбензимидазола и его сополимеров. По своим физико-механическим и адгезионным свойствам отечественные волокна до сих пор превосходят арамидные волокна Кевлар на основе поли п-фенилентерефталамида, разработанные в США фирмой Дюпон.
Волокна СВМ и Кевлар широко используются для армирования пластиков, применяемых в различных отраслях современной техники. С появлением жесткоцепных полимеров началось интенсивное изучение молекулярных композитов, в которых армирующая фаза из жесткоцепного полимера диспергирована на молекулярном уровне и равномерно распределена в матрице из гибкоцепного полимера, что позволяет создавать прочные и жесткие полимерные материалы при небольшом содержании жесткоцепного компонента.
Основная проблема при создании молекулярных композитов через растворы в общем растворителе заключается в совместимости и фазовой стабильности смесей жесткоцепных и гибкоцепных полимеров при сохранении высокодисперсного состояния фибрилл из жесткоцепного полимера при осаждении в твердой фазе. Эту проблему до сих пор пытались решить путем улучшения совместимости компонентов за счет введения дополнительных компонентов-«совместителей» (как правило, сополимеров с гибкими и жесткими фрагментами).
Нами предложен иной подход, позволяющий управлять процессом формирования морфологии композитов при формовании и осаждении из растворов смесей полимеров, реализуемом в технологии получения волокон из раствора. В условиях течения смеси полимеров через тонкий капилляр (фильеру) возникают сдвиговые и растягивающие напряжения, способствующие формированию в дисперсной фазе ультратонких волокон из жесткоцепного полимера. В случае полиамидбензимидазола, при термообработке композита выше температуры стеклования, происходит ЖК-переход с самопроизвольной ориентацией макромолекул, сопровождаемый значительным возрастанием прочности и жесткости микроволокон, без дополнительного воздействия механического поля.
Используя кинетически устойчивые растворы смесей промышленно выпускаемых жесткоцепных и гибкоцепных полимеров, удалось по простой и доступной технологии создать композиты с регулируемыми прочностными и термодеформационными свойствами, в том числе термопластичные теплостойкие пластики.
Работа выполнена при финансовой поддержке РФФИ (Грант № -а) и Программы ОХНМ РАН (проект № ОХНМ-03/128-117/ ).
МАТЕМАТИЧЕСКОЕ МОДЕЛИРОВАНИЕ ОДНОСТАДИЙНОГО ПРОЦЕССА СИНТЕЗА ПОЛИИМИДОВ В «АКТИВНОЙ» СРЕДЕ
А1., 1, 2
1Институт синтетических полимерных материалов им. РАН
2
Как показано нами ранее, расплав бензойной кислоты (БК) может быть использован в качестве среды вместо высококипящих растворителей (нитробензол, м-крезол) для получения полиимидов высокотемпературной поликонденсацией диаминов и диангидридов ароматических тетракарбоновых кислот. Преимуществами использования БК являются снижение температуры и продолжительности синтеза, а также отсутствие токсичности растворителя. В настоящей работе проведено математическое моделирование данного процесса. Использовали кинетическую схему, включающую основные реакции: полиацилирование и имидизацию промежуточно образующейся полиамидокислоты (ПАК), побочные реакции гидролиза ангидридных звеньев и имидных циклов выделяющейся водой, а также удаление воды. Константы скорости соответствующих реакций были получены в независимых экспериментах при изучении модельных реакций в ледяной уксусной кислоте.
Процедура включает: 1) составление системы ДУ, отражающих изменение концентраций всех компонентов системы с учетом уравнений материального баланса; 2) подстановку в ДУ начальных условий и соответствующих (констант скоростей модельных реакций, эксттраполированных к температуре синтеза ПИ, 3) численное интегрирование полученной системы ДУ с использованием математического пакета Maple 10.
Решение системы ДУ получено в виде графиков изменения во времени концентраций аминогрупп, ангидридных и орто-карбоксиамидных групп, фрагментов фталевой кислоты, а также изменения изменения среднечисленной степени полимеризации. Установлено, что концентрация орто-карбоксиамидных групп достигает максимума уже через несколько секунд и становится близка к нулю через 10 минут после начала реакции. Скорость процесса на всем протяжении процесса контролируется реакцией имидизации. Гидролиз ангидридных групп обратим и практически не оказывает влияния на процесс. Предельно достижимая молекулярная масса определяется обратимым характером всего процесса в целом. Графики изменения Рn во времени очень чувствительны к режиму удаления воды и к содержанию «условно неудаляемой» воды. В целом, получено хорошее качественное соответствие между расчетными и экспериментальными данными. Компьютерный анализ позволил объяснить ряд необычных закономерностей процесса, таких, например, как слабое влияние основности исходных диаминов на общую скорость, а влияние химического строения и концентрации мономеров на конечную молекулярную массу, а также, в ряде случаев, выбрать оптимальный режим проведения процесса.
Литература
1. , , Высокомолек. соед. А. 2007. Т.49. № 11. С. 1895.
2. Кuznetsov A. A., Tsegelskaya A. Yu., Buzin P. V., Yablokova M. Yu., Semenova G. K., High Performance Polymers. 2007. V.19: P.711.
ТРАНСПОРТ ИНФОРМАЦИИ ЧЕРЕЗ МЕМБРАНУ
, ,
Роль мембранного разделения отмечается в работах Тимашева [1,2]. В настоящей работе рассматривается имитационная модель эволюции частиц в двух боксах, разделенных мембраной, содержащих разные концентрации ионов. Мембрана содержит поры, размер и распределение которых задается исследователем [3]. В начальный момент в каждом боксе присутствуют незаряженные частицы, частицы с зарядом +3 и с зарядом -2, причем в таком соотношении, которое удовлетворяет принципу равенства зарядов. Эксперимент заканчивается после достижения теплового равновесия, которое характеризуется временами релаксации. В [4,5] приводится формулы, связывающие ионную силу I в модели Дебая-Хюккеля со свободной энергией F=(-e2η/6ε)*I, где ε – диэлектрическая проницаемость среды, η – обратный радиус ионной атмосферы, Т – температура. Отсюда получается соотношение, связывающее ионную силу и энтропию: 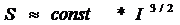 . Ниже приведены графические зависимости S1/S2 от I1/I2 для левого и правого бокса:
. Ниже приведены графические зависимости S1/S2 от I1/I2 для левого и правого бокса:
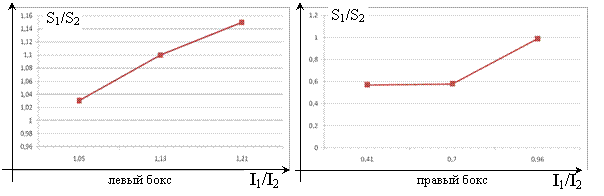

Рис. 1 Зависимость S1/S2 от I1/I2 для левого и правого боксов
Сравнивая ионные силы до опыта и после опыта можно оценить изменение энтропии. Соответственно, можно оценить величину передачи информации при смешении разных растворов различных концентраций.
Из полученных результатов видно, что с уменьшением значения I1/I2 пропорционально уменьшается и количество энтропийной информации, что указывает на статистический характер энтропии, т. е. нельзя присвоить какое-то количество «квантов» энтропийной информации конкретной индивидуальной частице. При уменьшении разности концентраций отклик системы и относительное количество переданной информации уменьшается. По изменению ионной силы до начала и после завершения опыта можно оценить, как далеко она находилась от того состояния, когда потоки достигают определенного порогового значения.
ЛИТЕРАТУРА
1. , Физикохимия мембранных процессов // М: Химия, 1988, 240 с.
2. // Электрохимия, 2006, т.42, c.480.
3. , , Эффект «Накачки» при диффузии через бесконечно тонкую мембрану // ДАН, т.354, №1, 1997, c.62-64.
4. Статистическая механика // М: «Комкнига», 2007, 448 с.
5. , Статистическая термодинамика в физической химии // М: БИНОМ, 2005, 495 с.
Математическое моделирование процесса
сорбции для сферических частиц
1, 2
1ГОУ ВПО «Южно-Уральский государственный университет» филиал в г. Златоусте
456209 Челябинская обл., 6
*****@***ru
2ГОУ ВПО «Московский государственный университет технологии и управления»
При изучении явлений массообмена в различных системах, достаточно часто возникают задачи, связанные с необходимостью рассчитать процесс сорбции, производимый сферическими частицами. Предлагается способ компьютерного моделирования процесса сорбции, позволяющий изучать его динамику.
При рассмотрении динамики процесса сорбции, производимого сферическими частицами можно выделить две стадии: диффузионный перенос субстрата к поверхности частицы и диффузионный перенос субстрата внутри частицы. Уравнения массообмена для выделенных стадий процесса имеют вид:
| (1) |
где D1, D2 – коэффициенты молекулярной диффузии субстрата для выделенных стадий процесса; L1, L2 – массовая концентрация субстрата, как функция времени и координаты для выделенных стадий процесса.
 Дополнив уравнения (1) начальными и граничными условиями, получим систему, описывающую процесс сорбции субстрата сферической частицей.
Дополнив уравнения (1) начальными и граничными условиями, получим систему, описывающую процесс сорбции субстрата сферической частицей.
Для анализа математической модели осуществлена её программная реализация на ЭВМ. Дифференциальные уравнения модели решались методом конечных разностей с использованием конечно-разностных аппроксимаций производных. Решение уравнений модели в этом случае позволит определить распределение концентрации сорбируемого субстрата внутри частицы в различные моменты времени. Пример полученного распределения концентрации в зависимости от времени представлен на рис. 1.
МАТЕМАТИЧЕСКОЕ МОДЕЛИРОВАНИЕ ТРЕХФАЗНЫХ ГЕТЕРОГЕННО-КАТАЛИТИЧЕСКИХ РЕАКТОРОВ
,
Федеральное Государственное Унитарное предприятие Научно-исследовательский физико-химический институт им , ,
*****@***ru
Многофазные гетерогенно-каталитические процессы и реакторы получили широкое распространение в промышленной практике. В настоящее время используют два типа реакторов: с орошаемым слоем катализатора, т. н. "trickle-bed reactor", и реакторы с суспендированным катализатором, т. н. “slurry reactor”. В trickle-bed реакторах используют катализаторы в виде гранул с характерным размером 2-5 мм, в slurry реакторах – тонкодисперсный катализатор размером 50-100 мкм.
Несмотря на широкое распространение, расчет и оптимизация многофазных реакторов вызывает большие затруднения, обусловленные сложностью аэрогидродинамики распределения потоков в объеме слоя и макрокинетики процессов переноса реагентов к поверхности зерна катализатора, диффузии реагентов в порах и химической реакции с выделением/поглощением тепла.
В настоящей работе методом математического моделирования на примере реакции гидрирования бензола в циклогексан на нанесенном катализаторе Pt на активном угле рассмотрены особенности макрокинетики гетерогенно-каталитической реакции в реакторах с орошаемым зернистым слоем и суспендированным катализатором. Кинетическая модель реакции разработана с сотрудниками [1].
Для описания реактора с орошаемым слоем разработана математическая модель, учитывающая межфазный массоперенос, диффузию и химическую реакцию в порах катализатора, сопровождающуюся выделением тепла, а также возможную неполноту смоченности зерна и изменение объема жидкой фазы в процессе реакции.
Разработана программа расчета и проведены расчетные исследования основных закономерностей процесса гидрирования бензола при различной плотности орошения и соотношении потоков газ-жидкость для зерен катализатора разного размера и разной глубины пропитки активным компонентом. Показано, что во всем исследованном диапазоне параметров режим работы реактора близок к внешнедиффузионному. Лимитирующей стадией массопереноса является скорость передноса растворенного водорода к внешней поверхности зерна катализатора. Показана целесообразность использования катализаторов корочкового типа, поскольку катализатору определенной активности отвечает оптимальная глубины пропитки зерна.
Проведено сравнение эффективности работы реакторов с орошаемым слоем и суспензионных. Показано, что при заданной конверсии бензола производительность реактора с орошаемым слоем в расчете на единицу массы катализатора значительно ниже, чем реактора с суспендированнным катализатором, а в расчете на единицу объема реактора их производительности близки.
Литература
1. , , Темкин жидкофазного гидрирования бензола на платиновом катализаторе // Кинетика и катализ. 1985. Т.26. №6. С.1489.
ДИСКРЕТНАЯ МОДЕЛЬ ПОЛЯРНОЙ ЖИДКОСТИ И ЭЛЕКТРОФИЗИЧЕСКИЕ ПРОЦЕССЫ С УЧАСТИЕМ ИЗБЫТОЧНЫХ И СОЛЬВАТИРОВАННЫХ ЭЛЕКТРОНОВ.
,
Федеральное государственное унитарное предприятие «Ордена Трудового Красного Знамени научно-исследовательский физико-химический институт имени », Москва, 105064 Воронцово поле 10, *****@***nifhi. *****
Разработана дискретная модель полярной жидкости, которая состоит из множества нейтральных и заряженных молекул с постоянными дипольными моментами. Учтена микроструктура полярной жидкости и установлена электростатическая неоднородность жидкости. В рамках дискретной модели вычислены локальные электрические поля и локальные электрические потенциалы на молекулах. Локальные потенциалы на любой молекуле строго индивидуальны и все время изменяются из-за движения молекул. Однако при этом сохраняются распределения молекул по локальным потенциалам и другие характеристики, через которые выражаются экспериментальные данные. Сформулированы основные положения теории, впервые объяснено большое количество экспериментальных данных для полярных жидкостей с избыточными и сольватированными электронами. Для описания свойств жидкости использована электронная волновая функция жидкости в виде произведения (Френкель, 1931) волновых функций молекул и ионов. Учтено, что молекула (без сродства к электрону) может на короткое время присоединить к себе электрон и образовать нестабильный анион (или резонанс) с определенной энергией и временем жизни. В результате избыточные электроны в жидкости все время находятся на какой-либо молекуле (в составе аниона-резонанса), а их перескоки с молекулы на молекулу описываются с помощью известной формулы Дирака для вероятности квантового перехода с поглощением или испусканием фотона. Эта формула, которая использовалась при расчете сил осцилляторов электронных переходов в атомах и молекулах, была применена нами для квантовой системы, коей является жидкость с множеством электронов в ней. В этой системе «движения» лишних электронов подчиняются законам квантовой механики; эти электроны не имеют кинетической энергии, они характеризуются функцией распределения по энергиям; при поглощении кванта света они «перескакивают» на соседние молекулы с большей локальной энергией; при перескоке электрона на молекулу с меньшей энергией фотон испускается. Теория объяснила основные характеристики жидкостей с сольватированными электронами: положение и форму оптического спектра поглощения; узкий спектр ЭПР электронов в воде; спектры фотоионизации сольватированных электронов в металл-аммиачных растворах; пороги фотоионизации (9.3 эВ) и внутренней фотоионизации (6.5 эВ) воды. Предсказаны спектры излучения жидкостей с избыточными электронами.
ЭЛЕКТРОННАЯ ПЛОТНОСТЬ СИСТЕМЫ He–(2S) И ЕЁ ПРИМЕНЕНИЕ ДЛЯ НАХОЖДЕНИЯ ПАРАМЕТРОВ РАССЕЯНИЯ.
,
Федеральное государственное унитарное предприятие «Ордена Трудового Красного Знамени научно-исследовательский физико-химический институт имени », Москва, 105064 Воронцово поле 10, *****@***nifhi. *****
Ранее авторами был развит метод нахождения автоионизационных состояний атомов и ионов с помощью эффекта стабилизации энергии этих состояний в вариационных расчетах с последовательно увеличивающимся базисом [1,2]. Однако, извлекаемая при этих расчетах информация об изучаемых системах гораздо обширнее и позволяет рассматривать процессы взаимодействия атома (или иона) с электроном в широком диапазоне энергий – от энергий тепловых электронов до энергий ионизации системы и выше. Была рассчитана 3-х-электронная система He–(2S). Построением графика радиальной электронной плотности ρ(r) с последующей его аппроксимацией при больших расстояниях от ядра r по формуле
![]()
была найдена зависимость от энергии для фазового сдвига δ0(E) или δ0(k) (E = k2/2). Существенным преимуществом метода является возможность определения δ0(E) при очень малых энергиях (E ~ 0,01 эВ), где существенную роль играют эффекты электронной корреляции и поляризации мишени рассеиваемым электроном. Анализ зависимости k·ctgδ0(k) от k в этой области энергий обнаруживает, по выражению Рамзауэра, «тонкую структуру», объяснение которой может быть дано только в модифицированной теории эффективного радиуса [3], которая явно учитывает дальнодействующую поляризационную часть потенциала
![]() (α – электронная поляризуемость атома),
(α – электронная поляризуемость атома),
что приводит к появлению в разложении k·ctgδ0(k) линейных (~ k) и логарифмических (~ ln k) членов.
1. , . Вычисление энергий резонансов в атомах и ионах с двумя и тремя электронами методом стабилизации. ЖФХ. 2007. т.81. №2. с.258-264.
2. , . Энергии резонансов в атомах и ионах с тремя электронами. ЖФХ. 2008. т.82. №3. с.460-464.
3. T. F. O'Malley, L. Spruch, L. Rosenberg. Modification of Effective-Range Theory in the Presence of a Long-Range (r–4) Potential. Journal of Mathematical Physics. 1961. V. 2. N.
ЗОННАЯ СТРУКТУРА УГЛЕРОДНЫХ НАНОТРУБОК, ИЗМЕНЕННАЯ В РЕЗУЛЬТАТЕ ПОВЕРХНОСТНОЙ АДСОРБЦИИ АТОМОВ ВОДОРОДА
,
Волгоградский государственный университет,
пр-т Университетский, 100, г. Волгоград, Россия
E-mail: *****@***ru, nikolay. *****@***ru
В работе представлены результаты теоретического квантово-статистического исследования адсорбции атомарного водорода на поверхности однослойных углеродных нанотрубок (УНТ)[1]. Использован модельный гамильтониан Андерсона [2,3]. В рамках метода функций Грина рассчитана зонная структура углеродных нанотрубок с адсорбированным атомом водорода.
![]()
где εk - энергия электров в идеальных УНТ, Vka – энергия гибридизациии адсорбированного атома (адатома), εa – энергия электрона в адатоме.
Проанализировано изменение зоны в результате атомарной адсорбции. Показано, что присоединение одного атома водорода уменьшает ширину запрещенной зоны полупроводящих углеродных нанотрубок. Качественное изменение зоны наблюдается в районе уровня адатома (примерно -10.6 эВ), расположенного в глубине валентной зоны. В этой области появляются дополнительные примесные уровни благодаря взаимодействию электрона атома водорода с валентной зоной кристаллической решетки.
Были рассмотрены также периодические дефекты двух видов: «квантовый провод», (атомы водорода располагались вдоль прямой, параллельной оси трубки) и «квантовый цилиндр», (атомы водорода адсорбировались на гексагональную сетку атомов углерода с различным периодом).
Построенная модель может быть использована для анализа изменения физических свойств углеродных нанотрубок в результате адсорбции одновалентных атомов. В построенной модели не учтены кулоновские корреляции электронов углеродных нанотрубок и колебания решетки.
Работа поддержана Российским фондом фундаментальных исследований (грант № ).
Литература:
1. Углеродные нанотрубы и родственные структуры. Новые материалы XXI века. Москва: Техносфера, 20с.
2. , Трошин на металлах и полупроводниках: модели Андерсона-Ньюнса и Халдейна-Андерсона // ФТТ. 2007. Т. 49. Вып. 8. С. 1508 – 1513.
3. , , Федорус пленки на поверхности металлов // УФН. 1977. Т. 122. Вып. 1. С. 125 – 158.
Математическое моделирование, оптимизация процесса и расчет реактора окислительной димеризации метана на лантан-цериевых катализаторах.
, ,
ГНЦ , 105064 , *****@***ru
В условиях сокращения мировых запасов нефти и резкого роста цен на нефтепродукты природный газ привлекает все большее внимание как альтернативный источник сырья для нефтехимических производств. Традиционный метод переработки метана предусматривает в качестве первой стадии конверсию метана в синтез-газ, которая осуществляется при высоком давлении и температуре и потому, является энергоемким и требует больших капитальных затрат. Более привлекательными являются одностадийные методы переработки, в частности, метод окислительной димеризации метана (ОДМ), который позволяет получить этилен в одну стадию.
В РГУ им. совместно с ИОХ РАН им. разработан эффективная катализатор на основе оксидов лантана и церия [1] и совместно с НИФХИ им. изучена кинетика и разработана феноменологическая кинетическая модель процесса [2], описывающая скорость основной реакции образования этилена и этана и побочных реакций – превращения метана в СО, СО2, монооксида в диоксид и последовательные реакции глубокого окисления этана, этилена:
1. ![]() ;
;
2. ![]() ;
;
3. ![]() ;
;
4. ![]() ;
;
5. ![]() ;
;
6. ![]() ;
;
7. ![]() .
.
На основе предложенной кинетической модели выполнена теоретическая оптимизация для реактора идеального вытеснения и исследованы зависимости конверсии кислорода и селективности образования этан-этилена от времени контакта, температуры и соотношения реагентов. В результате исследований определены теоретически оптимальные условия, при которых достигается максимальная селективность по этилену. Установлено, что зависимость селективности реакции от температуры носит экстремальный характер - существуют низкотемпературная и высокотемпературная области значений температур, при которых достигается максимум селективности по этан-этилену, разделенные областью значений температуры, при которых селективность минимальна. На основе полученных закономерностей определены оптимальные условия проведения процесса и выполнен расчет реактора.
1. , , Докл. АН, 2001, т. 380, № 6, с. 791—794.
2. , , РХЖ, в печати.
ФИЗИКО-ХИМИЧЕСКАЯ МОДЕЛЬ ВОЗНИКНОВЕНИЯ ПЛАНЕТ СОЛНЕЧНОЙ СИСТЕМЫ: ПРИЧИНЫ РАЗЛИЧИЯ ХИМИЧЕСКОГО СОСТАВА ПЛАНЕТ
*, **
*Научно-исследовательский физико-химический институт им. , Москва ул. Воронцово поле 10; E-mail: *****@***nifhi. *****
**Институт физики атмосферы им. РАН, Москва Пыжевский пер. 3; E-mail: *****@***ru
Адекватные представления о механизме возникновения планет Солнечной Системы важны не только сами по себе как решение одной из фундаментальных загадок мироздания, но они могут стать ключом для раскрытия таких тайн природы как происхождение локализаций отдельных химических элементов в земной коре, происхождение жизни, современной атмосферы Земли, и т. д. Только поняв механизм возникновения планет, мы сможем правильно оценить объем жизненно важных для человечества минеральных ресурсов, которыми мы располагаем на Земле и которые мы можем ожидать найти на других планетах. Национальный исследовательский совет США (National Research Council) первой среди десяти главных проблем геологии и планетологии назвал в начале этого года проблему о том, как сформировались Земля и другие планеты и почему планеты (включая их атмосферы), образовавшиеся из единого небулярного облака, значительно отличаются одна от другой.
В работе излагается феноменологическая гипотеза, предлагающая оригинальное решение этой проблемы.
Мы принимаем точку зрения большинства планетологов мира о том, что Солнечная Система возникла в результате деградации небулы (гигантского облака плазмы), возникшего в результате взрыва Сверхновой около 4,6 млрд лет назад.
Вместе с тем, описывая механизм расширения и последующего сжатия небулы, который привел к формированию современных объектов Солнечной Системы, мы учитываем новейшие исследования, выполненные с помощью межпланетных кораблей и телескопов, в частности, результаты фотографирования процессов зарождения планетных систем телескопами, расположенными в космосе.
Мы впервые принимаем во внимание, что водород (атомы Н и Не составляли более чем 99% атомов небулы) хемосорбируется на пылинках и агломератах почти любого химического состава вследствие особых свойств его атомов и молекул и что именно это свойство водорода играло важнейшую роль в генезисе формировавшихся небесных тел.
В предлагаемой нами гипотезе развиты органично связанные представления о холодных физически формировавшихся объектах (ФФО) и теплых химически формировавшихся объектах (ХФО) Солнечной Системы и на базе этих представлений дано объяснение различий планет по химическому составу и агрегатному состоянию. На основе гипотезы сделаны осторожные предсказания преимущественного состава ядер холодных небесных тел, дано объяснение примерному постоянству средней плотности Меркурия, Венеры и Земли, низкой плотности холодных планет-гигантов и повышенной плотности занептуновых небесных тел, преимущественному концентрированию воды (льда) в замарсовой области и некоторым другим особенностям Солнечной Системы.
Гипотеза объясняет образование залежей СН4, в том числе гидрата метана, и других углеводородов на Земле как результат взаимодействия внутри земной коры между Н2, десорбировавшимся из пород, и СО2, образовавшимся при разложения карбонатов.
Гипотеза опубликована и докладывалась в России и за рубежом.
МОДЕЛЬНОЕ ОПИСАНИЕ ПРОЦЕССОВ ВОЗНИКНОВЕНИЯ ЖИВОЙ МАТЕРИИ: LOH-ГИПОТЕЗА
**, *
*Научно-исследовательский физико-химический институт им. , Москва ул. Воронцово поле 10; E-mail: *****@***nifhi. *****
**Институт физики атмосферы им. РАН, Москва Пыжевский пер. 3; E-mail: *****@***ru
Развиты представления о явлениях и процессах, которые привели к образованию гидрата метана и трансформации первичной Н2/He атмосферы в О2/N2 атмосферу на Земле в течение Архейского периода. Обсуждение проводится в контексте развиваемой авторами гидратной гипотезы возникновения простейших элементов живой материи (Life origination hydrate hypothesis (LOH-гипотеза)). Формирование Земли как одной из планет Солнечной Системы и образование на ней гидратов метана и других углеводородов, простейших элементов живой материи и современной атмосферы рассматривается как звенья неразрывной цепи термодинамически обусловленных неизбежных явлений и процессов, которые регулировались универсальными физическими и химическими законами. Приведены термодинамические оценки, показывающие принципиальную возможность реализации в природе различных компонентов гипотезы. Сделаны предположения о причинах существующего распределения основных химических элементов между объектами Солнечной Системы.
Развита и обсуждается оригинальная гидратная гипотеза возникновения простейших элементов живой материи (Life Origination Hydrate Hypothesis; LOH-hypothesis), включающая представления о взаимозависимости и взаимообусловленности процессов возникновения жизни, трансформации первичной атмосферы и формирования залежей гидрата метана. Впервые учитывается, что молодая Земля была "пропитана" небулярным водородом. Возникновение простейших элементов живой материи рассматривается как результат термодинамически обусловленных закономерных и неизбежных химических превращений и универсальных физико-химических законов. Согласно гипотезе, простейшие элементы живой материи многократно образовывались и, возможно, образуются из метана (или другого углеводорода), селитры и фосфата в пограничных областях твердых фаз газовых гидратов простейших углеводородов. Предполагается, что явление монохиральности нуклеиновых кислот является следствием особенностей геометрии структурной матрицы.
Развиты представления о явлениях и процессах, которые привели к образованию гидрата метана и трансформации первичной Н2/He атмосферы в О2/N2 атмосферу на Земле в течение Архейского периода. В качестве исходного положения принято мнение многих исследователей, которые полагают, что Солнечная Система возникла из небулы, образовавшейся при взрыве Сверхновой около 5 млрд лет назад. Обсуждение механизма рассматриваемых явлений и процессов проводится в контексте развиваемой гидратной гипотезы возникновения простейших элементов живой материи (Life origination hydrate hypothesis (LOH-гипотеза)). Формирование Земли как одной из планет Солнечной Системы и образование на ней гидратов метана и других углеводородов, простейших элементов живой материи и современной атмосферы рассматриваются как звенья неразрывной цепи термодинамически обусловленных неизбежных явлений и процессов, которые регулировались универсальными физическими и химическими законами. Приведены термодинамические оценки, показывающие принципиальную возможность реализации в природе различных компонентов гипотезы. Сделаны предположения о причинах существующего распределения основных химических элементов между объектами Солнечной Системы. В рамках развиваемой гипотезы рассматривается возможность существования жизни на различных планетах Солнечной системы. Обсуждаются выявленные в последние годы на Земле и в космическом пространстве природные явления и процессы, которые свидетельствуют в пользу развиваемой гипотезы. Проводится сопоставление LOH-гипотезы с другими опубликованными гипотезами мироздания.
We believe that the processes that led to living-matter origination and to its subsequent development are thermodynamically conditioned, natural, and inevitable and that all they are governed by universal physical and chemical laws.
According to the Life Origination Hydrate Hypothesis (LOH-hypothesis) [1–7], the living matter simplest elements (LMSE) originated and, possibly, originate in our days from CH4 (or other CH4-hydrocarbons), niters, and phosphates under the Earth's surface or seabed within honeycomb structures of hydrocarbon hydrates. The underground deposits of CH4 and other hydrocarbons could be produced by the reaction between H2 and CO2 and CO2 could be produced from carbonates as a result of their thermal decomposition induced by the gravitational compression of the young-Earth crust. Thus, the living-matter sources are H2, carbonates, and phosphates resulted from transformation of the nebula. The nebula that was the progenitrix for the Solar System arose after the supernova explosion.
The hypothetical sequence of the processes that led to formation of protocells from methane, niter, and phosphate is as follows: niter diffusion into methane hydrate structure → formation of N-bases and riboses within structural cavities → phosphate diffusion from outside into small structural cavities → formation of DNA - (RNA-) like molecules through polymerization → melting of the system and water–organic-soup formation → formation of amino-acids and simplest organelles in the soup → self-replication of nucleic acids and concentrating of the soup → formation of protocells.
The LOH-hypothesis allows for answering the following questions. From what substances and by what mechanism had the LMSE originated? How had it happened that methane hydrate had formed? How had it happened that CH4 and NO3– met together, and why had no other substances reacted with them? Why are the DNA and RNA monomer links limited in size, and why are they so similar? What had hampered the subsequent chemical transformations of the rings and side groups of N-bases and riboses? How had it happened that the sequences of N-bases in DNA and RNA molecules are not random? Why do atoms other than C, N, P, O, and H almost never enter the DNA and RNA compositions? Why do only five N-bases usually participate in DNA and RNA formation, and why do some other N-bases, e. g., xanthine, sometimes enter the DNA and RNA compositions? Why did Nature select the D-ribose molecules for DNA and RNA construction?
This presentation will contain the experimental and observational facts that led us to the formulation and development of the LOH-hypothesis, thermodynamic estimations showing that the LMSE can be produced on the basis of the internal energy of the source substances with no external heat flows, answers to the above-listed questions, available independent environmental observations counting in favor of the hypothesis, and descriptions of the experiments that could be performed for its validation and subsequent development.
ГРАФ РЕАКЦИОННЫХ МАРШРУТОВ КРОСС-МЕТАТЕЗИСА ОЛЕФИНОВ
, Illie Fishtik
НИФХИ им. , 0 *****@***nifhi. *****
Worcester Polytechnic Institute, Massachusetts, USA
Изменение реакционной способности α-олефинов при метатезиса на рений-содержащих гетерогенных катализаторах в многокомпонентных смесях получает естественное объяснение, если допустить, что их адсорбция протекает не диссоциативно, а путем вытеснения. Такая схема при определенных допущениях не позволяет получить кинетическое уравнение в явном виде, т. е. исключить все интермедиаты. В этом случае приходится использовать саму схему в качестве микрокинетической модели и находить из расчета и эксперимента значения кинетических параметров всех стадий. Построение графа реакционных маршрутов (ГРМ) существенно облегчает работу с микрокинетической моделью, поскольку обеспечивает не только ее наглядную визуализацию, но и возможность производить прямые количественные оценки.
Метод основан на фундаментальном соотношении Т. деДонде, точнее на его линеаризованном приближении (выражение справа)
![]()
Здесь rρ - скорость ρ-ой стадии, rρ+ - то же только в прямом направлении,
- химическое сродство A в приведенном виде,![]() Rρ - некая величина, придающая выражению для скорости элементарной стадии вид закона Ома.
Rρ - некая величина, придающая выражению для скорости элементарной стадии вид закона Ома.
ГРМ строится таким образом, чтобы он имел полную аналогию с электрической схемой, к которой были бы применимы оба правила Кирхгофа и закон Ома. При этом аналогом силы тока будет скорость реакции, а электрическое напряжение – эквивалентом химического сродства, что позволит использовать теорию электрических цепей для анализа схем механизма химических реакций.
Схема механизма кросс-метатезиса 1-октена и 1-децена была несколько упрощена. Электротехнический эквивалент ГРМ для неё приведен ниже, номера резисторов соответствуют номерам элементарных стадий.
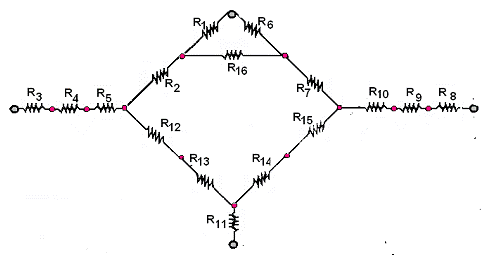
Со слайдами доклада можно ознакомиться по ссылке:
http://*****/conference/graph_doc. ppt
СИСТЕМА МЕНЕДЖМЕНТА КАЧЕСТВА РАДИАЦИОННЫХ ИСПЫТАНИЙ НЕМЕТАЛЛИЧЕСКИХ МАТЕРИАЛОВ
1, 2, 3, 4, 4
(1) », г. Москва
(2) », г. Обнинск Калужской области
(3) ГОУ ВПО «РХТУ им. », г. Москва
(4) Центр фотохимии РАН, г. Москва
Решение проблемы прогнозирования радиационной стойкости (РС) радио - и оптоэлектронных, электротехнических и других изделий на основе неметаллических материалов, используемых в полях ионизирующих излучений (эта ситуация возникает в космосе, ядерных энергетических установках (ЯЭУ) и т. д.), вызывает необходимость в организации и проведении радиационных испытаний (РИ).
Обычно рассматривают два вида РИ – натурные и лабораторные. Натурные РИ проводят в условиях, полностью соответствующих радиационной обстановке, в которой осуществляется эксплуатация материала. Однако этот вид РИ является трудоёмким, требует значительных финансовых затрат. Часто отсутствует возможность в организации и проведении РИ из-за технической недоступности области пространства, в которой необходимо проведение натурных РИ, или других причин (например, запрет на проведение испытаний ядерного оружия).
Лабораторные (наземные) РИ позволяют получить оценку РС на основании изучения закономерностей радиационного поведения неметаллических материалов, при воздействии стандартных видов ионизирующих излучений. Как правило, в качестве стандартного вида ионизирующего излучения (ИИ) при проведении лабораторных РИ используют γ-излучение изотопа 60Со со средней энергией 1,25 МэВ. Очевидно, в условиях лабораторных РИ можно лишь в незначительной степени моделировать реальную радиационную обстановку (мощность дозы, поглощённая доза, флюенс). Поэтому, лабораторные РИ не могут дать достоверную оценку РС. Следовательно, необходимо построение теоретических моделей, с помощью которых можно получить прогнозные оценки РС материала в реальных условиях эксплуатации. Проблема моделирования радиационных эффектов в неметаллических материалах является комплексной, и включает необходимость:
- сопоставления воздействия различных видов ИИ, различающихся плотностью ионизации (низкоэнергетические электроны и протоны радиационных поясов Земли, галактическое и космическое излучение, гамма-нейтронное излучение ЯЭУ, излучение ускорителей частиц, гамма-установок, рентгеновских аппаратов и т. д.);
- оценки вклада в радиационно-индуцированное изменение свойств исследуемого материала вторичных и побочных процессов (газовыделение, процессы деструкции и сшивания, радиолюминесценция, радиационно-химическое окисление и т. п.).
Таким образом, основными критериями качества РИ следует считать уровень имитации реальной радиационной обстановки в лабораторных условиях и надёжность прогнозных оценок РС изучаемых неметаллических материалов. Создание системы менеджмента качества РИ неметаллических материалов включает необходимость выполнения ряда научно-технических и организационных мероприятий, направленных на изучение механизмов РС неметаллических материалов, создание технической базы для проведения РИ, организацию и проведение РИ различных классов материалов, разработку нормативно-технической документации (НТД), регламентирующей порядок проведения РИ, а также подготовку кадров и широкое внедрение методов информационных технологий в практику РИ.
ФЛИККЕР-ШУМОВАЯ СПЕКТРОСКОПИЯ КАК НОВАЯ ИНФОРМАЦИОННАЯ ТЕХНОЛОГИЯ: ПРИНЦИПЫ И ПРИЛОЖЕНИЯ
Научно-исследовательский физико-химический институт им. ,
Москва, ; *****@***nifhi. *****
Фликкер-шумовая спектроскопия (ФШС) [1-4] – феноменологическй метод извлечения информации, содержащейся в хаотических сигналах, продуцируемых открытыми сложными, в том числе, природными системами разной сущности. Сущность ФШС состоит в придании информационной значимости нерегулярностям анализируемых сигналов – всплескам, скачкам, изломам производных различных порядков на каждом пространственно-временном уровне иерархической организации исследуемых систем и в установлении корреляционных взаимосвязей в последовательности фиксируемых значений исследуемых динамических переменных. Основное отличие ФШС от иных методов анализа хаотических сигналов состоит в введении информационных параметров, характеризующих составляющие исследуемых сигналов в разных диапазонах частот, и реализации необходимых процедур для выделения таких параметров. Согласно ФШС методологии, индивидуальные особенности эволюции сложных систем проявляются, прежде всего, в низкочастотных составляющих продуцируемых сигналов, отражающих специфический для каждой системы набор собственных и инициируемых сторонними воздействиями частот, интерференционные вклады таких резонансов. На фоне такого типа низкочастотных «огибающих» неизбежно присутствуют более высокочастотные хаотические («шумовые») составляющие, генезис которых далеко не всегда ясен, но в последовательности которых практически всегда выявляются высоко индивидуальные для каждой системы, информационно значимые корреляционные взаимосвязи. Поэтому лишь при последовательном выделении вкладов таких составляющих сложных сигналов в разных диапазонах частот и введении соответствующей параметризации возможно наиболее адекватно оценивать состояние исследуемой системы, динамику изменения состояния ее подсистем, в том числе, в условиях сторонних воздействий. При реализации соответствующих процедур возможно выявление хаотических составляющих, временные изменения которых можно описывать как процессы аномальной диффузии. ФШС метод эффективно используется для решения проблем 3-х типов: выявления параметров, характеризующих динамику открытых сложных систем; определения прекурсоров наиболее резких изменений в эволюции таких систем; установления динамики кросс-корреляционных взаимосвязей между одновременно измеряемыми характеристиками систем. Возможности ФШС методологии демонстрируются на примерах приложений к задачам физики, астрофизики, геофизики, электрохимии, медицины.
1.Тимашев -шумовая спектроскопия: информация в хаотических сигналах. М.: Физматлит. 20с.
2.Тимашев значимость хаотических сигналов: фликкер-шумовая спектроскопия и ее приложения. Электрохимия. 2006. Т.42. №5. С. 480-524
3. Timashev S. F., Polyakov Yu. S. Review of flicker noise spectroscopy in electrochemistry. Fluctuation and Noise Letters. 2007. V. 7. N. 2. P. R15-R47.
4. Timashev S. F., Polyakov Yu. S. Analysis of discrete signals with stochastic components using flicker noise spectroscopy. International Journal of Bifurcation and Chaos. 2008. V. 18. N 9.
5. Timashev, S. F. and Polyakov, Yu. S. A review of flicker-noise spectroscopy: information in chaotic signals. In: Simultaneity: Temporal Structures and Observer Perspectives. Eds. Susie Vrobel, Otto E. Rössler and Terry Marks-Tarlow (Festschrift: 10th anniversary of the Institute for Fractal Research, Kassel, Germany). Singapore: World Scientific, 2008. P. 270-285
МОДЕЛИРОВАНИЕ ТРАНСПОРТА МОЛЕКУЛ
В УЗКИХ ЩЕЛЕВИДНЫХ ПОРАХ
, ,
ГНЦ РФ “НИФХИ им. Л.Я. Карпова”, Москва, Воронцово Поле 10, *****@***nifhi. *****
Стадия транспорта молекул в узких каналах присутствует во всех молекулярных процессах в пористых телах. Потенциал поверхностных сил создает сильно анизотропное распределение молекул в сечениях узких порах. Аналогичное сильно анизотропное распределение молекул возникает при наличии границы раздела паровой и жидкой фаз в случае капиллярной конденсации. В обоих случаях уравнения гидродинамики неприменимы. Обсуждаются основы кинетических уравнений, описывающих перенос молекул в узких порах в области капиллярных явлений, когда необходимо иметь единую систему уравнений, описывающую течения, как плотных газов, так и жидкостей.
Вопрос о типе уравнений, описывающих динамику движения мениска на границе пар - жидкость остается неясным до настоящего времени. При рассмотрении транспорта пара или жидкости в каналах заданного сечения любого малого элемента пор имеется четкая аналогия со стандартными уравнениями гидродинамики. При переходе к макроскопической системе пор в целом, процессы транспорта молекул описывают диффузионными уравнениями, связывающими поток массы с общим перепадом химического потенциала по обе стороны твердого тела.
Для расчета молекулярных потоков использован новый микро-гидродинамический подход, основанный на простейшей молекулярной модели - модели решеточного газа, которая учитывает собственный объем молекул и межмолекулярные взаимодействия в квазихимическом приближении. Новый подход позволяет рассматривать каналы в нанометровом диапазоне от 1 до 100 нм.
Построенная система уравнений является системой в конечных разностях по приращениям координат (вместо дифференциальных производных). Диссипативные коэффициенты учитывают нелокальные свойства флюида.
Модель охватывает изменения концентраций флюида от газообразного до жидкого состояния и широкий диапазон температур, включая критическую область, что позволяет рассматривать динамику течений пара, жидкости и паро-жидкостных флюидов при наличии капиллярной конденсации. При увеличении размера пор, полученные уравнения переходят в гидродинамические уравнения переноса для потоков газа или жидкости, сохраняя связь коэффициентов переноса с межмолекулярными потенциалами.
Обсуждается влияние потенциала адсорбент – адсорбат на коэффициенты диффузии и сдвиговой вязкости, а также на характеристики молекулярных потоков в узких щелевидных порах.
Исследованы динамические режимы течения одноатомного газа (аргона) в щелевидных порах разной ширины при разных способах возмущения начального равновесного распределения молекул, которое рассчитывается с учетом переменной плотности по сечению поры.
Показано, что при сильном притяжении атомов аргона малой плотности к стенкам поры преимущественно наблюдается пленочное течение. Уменьшение притяжения молекул к стенкам приводит к реализации режима проскальзывания флюида вдоль стенки канала.
Работа выполнена при поддержке РФФИ (проект № а).
СТИМУЛИРОВАННАЯ ПОВЕРХНОСТНАЯ СЕГРЕГАЦИЯ ХИМИЧЕСКОГО СОСТАВА: ТЕОРИЯ, ЭКСПЕРИМЕНТ
, .
»
Дан обзор теоретических и экспериментальных результатов в области стимулированной температурой или иными факторами поверхностной автосегрегации химического состава оксидов (СПАС) сложных оксидов. Движущая сила СПАС – уменьшение свободной энергии при изменении поверхностного химического состава.
Модельные представления предполагают диффузионный механизм сегрегации, параметры теории содержат упругие константы и кристаллохимические характеристики веществ. Все исследованные вещества разделены на группы с различной степенью сегрегации. На основе моделей можно предсказать характер изменений поверхностного состава оксидов: тип сегреганта и степень сегрегации.
При СПАС на поверхности матрицы – подложки может быть сформирован слой (пленка) соединения (или твердого раствора) из компонентов матрицы, который будет иметь структуру и состав, отличные от матрицы. Достоинства синтеза на основе СПАС заключаются в высокой адгезии пленок, возможности получения пленок нанометрического масштаба толщин, возможности эпитаксиального роста (новый вид эпитаксии), возможности создания функциональных слоев, например, для эмиссионной наноэлектроники (рис.1), слоевых систем с различным сочетанием функциональных свойств (сегнетоэлектрик-диэлектрик, полупроводник, сверхпроводник-полупровод-ник и др.)
СПАС исследована в монокристаллах: PbTiO3 , SrTiO3 , Pb Fe 0,5 Nb 0,5 O3 , BaTiO3, Bi4Ti3O12, LiNbO3, Pb 5 Ge 3 O11, Gd2 (MoO4)3, KNbO3 , YBa2Cu3O7 и керамике Pb(Ti, Zr)O3.
Монокристаллы PbTiO3 , BaTiO3 использованы в качестве матриц - подложек для получения поверхностных эпитаксиальных слоев TiO2 и BaTi2O5.
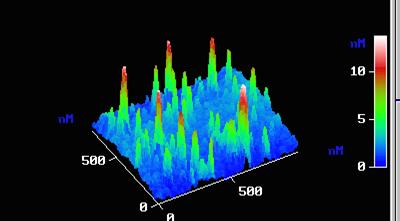
Рис.1. Ансамбль наночастиц TiO2 высотой 5-10 нм на поверхности кристалла PbTiO3
ИСПОЛЬЗОВАНИЕ ЭНЕРГИИ ХИМИЧЕСКИХ РЕАКЦИЙ
Федеральное государственное унитарное предприятие «Ордена Трудового Красного Знамении научно-исследовательский физико-химический институт имени Л. Я. Карпова», Москва, Воронцово поле, д. 10, *****@***ru.
В работе рассмотрена возможность рационального использования энергии химических реакций, с учётом требований второго закона термодинамики, и в том числе принципа Ле Шателье и принципа противодействия, применительно к классической задаче увеличения выхода продуктов и задаче снижения затрат энергоресурсов, с помощью увеличения потенциала энергии экзотермической реакции. За счёт увеличения потенциала энергии химической реакции появляется возможность использовать продукты химической реакции в качестве рабочего тела в схеме с турбиной, с получением помимо целевого продукта ещё и электроэнергии. В частности проанализированы процессы синтеза метанола, аммиака, триоксида серы, α-метилстирола и производство синтез-газа высокотемпературной конверсией метана.
Рассмотрено соотношение затрат энергии на компрессию газа и компенсация их за счёт энергии расширяющихся продуктов реакции. На основе проделанной работы разработаны требования к процессам, для которых возможно использовать энергию химических реакций по предложенной схеме.
Теоретическое изучение процесса образования аци-формы в ряду динитроалканов
, ,
Казанский Государственный Технологический Университет, Казань,
ул. К. Маркса, д. 68, а/я 170, *****@***ru
Изучение таутомерных превращений представляет значительный интерес для понимания общих закономерностей реакционной способности нитросоединений. С использованием различных теоретических и экспериментальных методов исследования доказана важная роль изомеризации с образованием нитроновых кислот в механизме термодеструкции нитроалканов в жидкой фазе. Вместе с тем механизм термодеструкции нитроалканов изучен недостаточно. Значительный интерес, в частности, представляет объяснение особого положения динитрометана, который отличается крайне низкой химической стойкостью, в то время как моно-, три - и тетранитроалканы в газовой фазе являются достаточно стабильными соединениями. Естественно предположить существование механизма термораспада, для которого динитрометан является особой системой.
Нами изучена реакция образования аци-формы в молекулах динитрометана, динитроэтана и динитропропана. Для данных реакций определены геометрические параметры переходных состояний и проведена оценка барьеров активации. Геометрическая структура переходного состояния для данной реакции приведена на рис. 1. Барьер активации для данной реакции составил 231.3 кДж/моль.

Рис. 1 Геометрическая структура переходного состояния для реакции образования аци-формы в молекуле динитрометана.
Теоретическое изучение проводилось с использованием программы Gaussian 98, DFT методом B3LYP c различным набором базисов.
Для молекулы аци-формы динитрометана проведен конформационный анализ, в ходе которого была определена наиболее стабильная структура данного соединения.
State-of-the-arts in the electron affinities calculations. Clusters of alkaline-earth elements
Ilya G. Kaplan
Instituto de Investigaciones en Materiales, Universidad Nacional Autónomo de México, AP.70-360, 04519 México, D. F., México
If in atoms only one type of the electron affinity (EA = E0 (N+1) – E0 (N)) can be defined, in molecules there are three types of EAs: the vertical electron affinity (VEA), adiabatic electron affinity (AEA), and vertical electron detachment energy (VEDE). For reliable calculation of EAs, the employment of high-level electron correlation methods are needed, for instance, the employment of the DFT approaches, even with the modern hybrid potentials, can lead to qualitatively wrong conclusions. The basis sets used for anion calculations have to contain diffuse functions with high angular momenta in order to describe the binding of remote excess electron. They also have to be flexible enough to describe large relaxation upon electron attachment. The Dunning-type augmented correlation consistent aug-cc-pVXZ basis sets with X = T, Q, etc. are the most appropriate.
The study of binding of an excess electron to clusters of alkaline-earth elements is interesting as an instructive example of unusual properties of complexes of atoms possessing closed electron shells. In Refs [1-3] all three types of EAs of the Ben and Mgn (n = 2-4) clusters were calculated at different levels of theory using the aug-cc-pVQZ basis sets. EAs of Be2 and Be3 were calculated up to the complete CCSDT level [1]. This benchmark calculation shows that the MP4(SDTQ) results overestimate and CCSD(T) approach slightly underestimates the magnitude of EA, whereas the global minimum in the potential energy surface becomes deeper with a greater accounting of the correlation energy in the row: MP4(SDTQ), CCSD(T), AND CCSDT .
In this comparative study we recalculated EAs for Be and Mg clusters at the MP4(SDTQ) level, taking into account all electrons (without the frozen core approximation) and calculated at the same level the Ca clusters. The obtained values of VEDE for all studied clusters are large enough to be observed with standard photodetachment technique. The decomposition of VEDE into three components (Koopmans, relaxation, and correlation) and the atomic orbital population analysis at the NBO level are used to elucidate the nature of the outer electron binding in studied anions.
1. Kaplan, I. G.; Dolgounitcheva, O.; Watts, J. D.; Ortiz, J. V. J. Chem. Phys. 2002, 117, 3
2. Kaplan, I. G. ; Diaz, C. C. Int. J. Quant. Chem. 2005, 104, 468-474.
3. Diaz, C. C.; Kaplan, I. G.; Roszak, S. J. Mol. Modeling, 2005, 11, 330-334.
