
Партнерка на США и Канаду по недвижимости, выплаты в крипто
- 30% recurring commission
- Выплаты в USDT
- Вывод каждую неделю
- Комиссия до 5 лет за каждого referral
1. Построение выравнивания последовательностей пары протеинкиназ с разметкой кластеров плюс-блоков.
Рассмотрим цепи В структур протеинокиназ 1AD5 и 1K9A.
Первый белок представляет собой тирозин-киназу Hck кроветворных клеток, второй – карбоксил-терминальную Src киназу.
Длины выбранных цепочек – 531 и 450 а. о. соответственно, длина выравниваемых цепей - 438 и 441 а. о. (причина лежит в отсутствии некоторых координат).
Данные цепочки были поданы на вход программе гибкого выравнивания FATCAT.
Некоторая информация о полученном выравнивании:
P-value = 1.11e-16,
RMSD = 2.72,
выравнивание содержит 2 изгиба, 416 эквивалентных позиций.
Оригинальное выравнивание находится в файле fatcat. txt в папке с заданием, далее же будет представлено выравнивание в формате msf, конвертированное с помощью скрипта fatcat_to_fasta. py, с выделенными консервативными позициями и кластерами:
Всего было найдено 3 кластера плюс-блоков.
Длина выравнивания – 446, суммарная длина обоснованного выравнивания – 416, процент от длины меньшей последовательности в выравнивании – 416/438 = 94,98%.
Сравним полученные кластеры:
Идентификатор кластера | Положение в выравнивании | Число плюс-блоков в кластере | Суммарное число позиций обоснованного выравнивания в кластере | Суммарное число совпадающих букв в кластере | Identity % | Суммарное число сходных букв в кластере | Similarity % | Мера сходства конформаций двух фрагментов из кластера блоков |
Block0 (A) | 1-33, 36-44, 46-63 | 3 | 60 | 18 | 30 | 31 | 51,7 | score 159.26 rmsd 1.38 |
Block1 (B) | 70-90, 93-117, 123-134, 136-175 | 4 | 98 | 32 | 32,65 | 41 | 41,8 | score 254.39 rmsd 1.89 |
Block2 (С) | 183-215, 217-226, 228-260, 263-339, 342-446 | 5 | 258 | 113 | 43,8 | 153 | 59,3 | score 639.72 rmsd 1.30 |
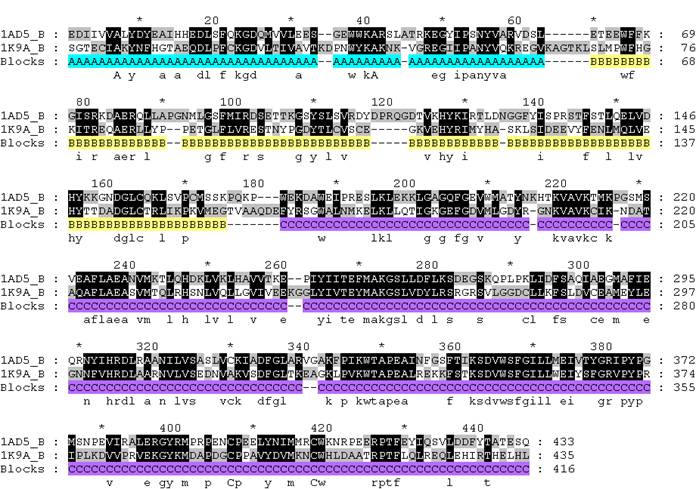
СОВМЕЩЕНИЕ одной структуры с изогнутой второй в PDB формате с раскрашенными кластерами плюс-блоков:
(где цепь А – структура 1ad5B, a цепь B – модифицированная структура 1k9aB)
и rasmol-скрипт 1AD5B.1K9AB. script
rasmol-скрипт для покраски кластеров преобразован в скрипт для PyMol:load 1AD5B.1K9AB. pdb
bg_color white
hide all
show cartoon
color gray
color palecyan, a/82-143/
color cyan, b/10-71/
color wheat, a/144-249/
color yellow, b/78-175/
color slate, a/254-526/
color marine, b/183-450/
ЗАКЛЮЧЕНИЕ
1. Хорошее совмещение кластеров плюс-блоков в пространстве (без видимых крупных погрешностей), низкое значение RMSD (<2), а также довольно высокий процент идентичности а. о. (>30) позволяют говорить о высокой степени доверия данному выравниванию.
Подозрения об ошибках программы в ходе выполнения работы не возникли.
2. Рассмотрим доменную организацию совмещаемых цепей (согласно pDomains):
Selected Protein: 1AD5 Chain: B
CATH 1AD5B1 1-63 1AD5B2 64-165 1AD5B3 176-261 1AD5B4 262-426
SCOP 1AD5B1 1-63 1AD5B2 64-166 1AD5B3 167-438
Selected Protein: 1K9A Chain: B
CATH 1K9AB1 6-67 1K9AB2 86-171 1K9AB3 187-272 1K9AB4 273-443
SCOP 1K9AB1 4-76 1K9AB2 77-177 1K9AB3 178-450
Границы доменов двух цепей незначительно отличаются от координат кластеров плюс-блоков и друг от друга,
эти различия в совокупности с делением методом CATH последнего домена на два указывают на некоторую степень изменчивости конформации рассматриваемых белков, что скорее всего говорит об их конформационной подвижности, также небольшой вклад могут вносить ошибки и погрешности кристаллизации, особенно на граничных участках кластеров.
Белок 1AD5 был получен из организма человека, а 1K9A - выделен из организма крысы, так что роль эволюционной изменчивости в изменчивости конформации невелика, против нее также говорят высокие проценты идентичности и сходства а. о. в цепях.
2 Построение гибкого выравнивания с помощью сервиса RAPIDO
для структур из упр.1.
Информация о полученном выравнивании (по ссылке можно посмотреть выравнивание с размеченными кластерами):
1st struct. 1AD5_B (438)
2nd struct. 1K9A_B (441)
#aligned 399
RMSD rigid 22.90
#rigid 351
RMSD flex 0.96
# rigid bodies 4
Идентификатор кластера | Размер | Координаты | RMSD |
1 | 191 | 1AD5_B: B267-B287, B289-B298, B316-B325, B335-B352, B357-B404, B407, B425-B464, B470-B483, B486-B514 1K9A_B: B195-B225, B242-B251, B263-B280, B285-B332, B335, B349-B388, B394-B407, B410-B438 | 0.86 |
2 | 83 | 1AD5_B: B146-B163, B170-B191, B197-B205, B210-B221, B224-B245 1K9A_B: B80-B97, B102-B132, B136-B147, B150-B171 | 0.84 |
3 | 55 | 1AD5_B: B84-B114, B116-B124, B126-B140 1K9A_B: B12-B42, B45-B68 | 1.04 |
4 | 22 | 1AD5_B: B262-B266, B306-B307, B310-B315, B326-B334 1K9A_B: B190-B194, B232-B233, B236-B241, B252-B257, B260-B262 | 1.68 |
Прежде всего стоит отметить очень низкое значение RMSD; а также наличие плюс-блока, состоящего всего из 1 а. о. (выделен жирным в таблице), причина скорее всего заключается в том, что соседние а. о. отсутствуют в структуре.
Далее следует заметить, что по сравнению с FATCAT кластеры содержат большее количество плюс-блоков (вплоть до восьми).
В отличии от FATCAT, RAPIDO обнаружил 4 кластера, для структуры 1K9A_B координаты первых двух кластеров сходны с FATCAT, третий кластер поделен на 2, причем, что самое интересное, расположение плюс-блоков этих двух кластеров чередуется.
Для структуры 1AD5_B координаты кластеров сильно различаются:
FATCAT | RAPIDO |
1-63 70-175 183-446 | 84-140 146-245 267-514 262-334 |
расположение кластеров, согласно RAPIDO, смещену в сторону N-конца последовательности, опять же присутствуют два кластера с чередующимися плюс-блоками.
3. Сравнение пары структур одного и того же белка с помощью гибкого выравнивания
Рассматриваемый белок - гемофор HasA из бактерии S. marcescens (записи 2cn4 и 1dk0, сравниваем цепи A), отвечающий за “кражу” бактерией железа из эритроцитов.
Результаты работы сервиса FATCAT:
длина выравнивания – 173 позиции,
RMSD = 0.47,
Последовательности полностью идентичны.
Координаты кластеров – 2-48; 50-174.
Изображение гибкого выравнивания в PyMOL:
(зеленым цветом раскрашена цепь 2cn4_A, желтым - 1dk0_A)
Структуры в пространстве совмещаются почти полностью, попробуем разобраться в причине их небольших отличий, она может заключаться в конформационной подвижности белка либо в ошибках кристаллизации.
1. Обе последовательности идентичны, каждая структура состоит из двух доменов, расположенных впритык друг к другу и занимающих практически всю цепь, так что вариабельность конформаций невелика
(по версии БД SCOP и CATH домен и вовсе один).
2. Проверим вторую версию.
Для начала рассмотрим записи по-отдельности, запись 1dk0 содержит 2 идентичных полипептидных цепи, связанные с 2 молекулами гема.
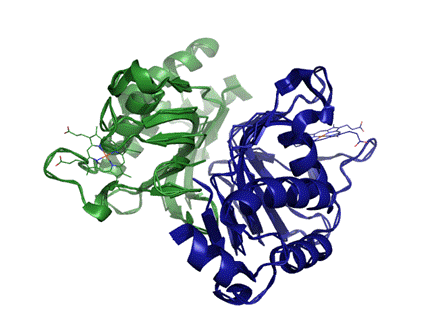
2cn4 имеет такую же структуру, но его цепи располагаются более тесно, переплетенно. Каждая молекула гема связана с обоими цепями.
Для проверки на предмет наличия ошибок кристаллизации поищем возможные контакты димеров и молекул гема внутри одной ассиметричоской единицы с белками и молекулами гема из других элементарных ячеек.
1dk0:
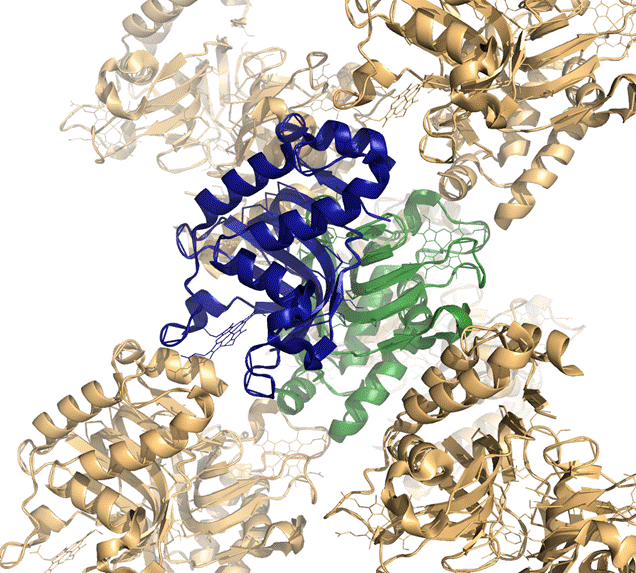
Каждая молекула гема связана с двумя цепями: из своей ассиметрической единицы и из соседней.
2cn4:
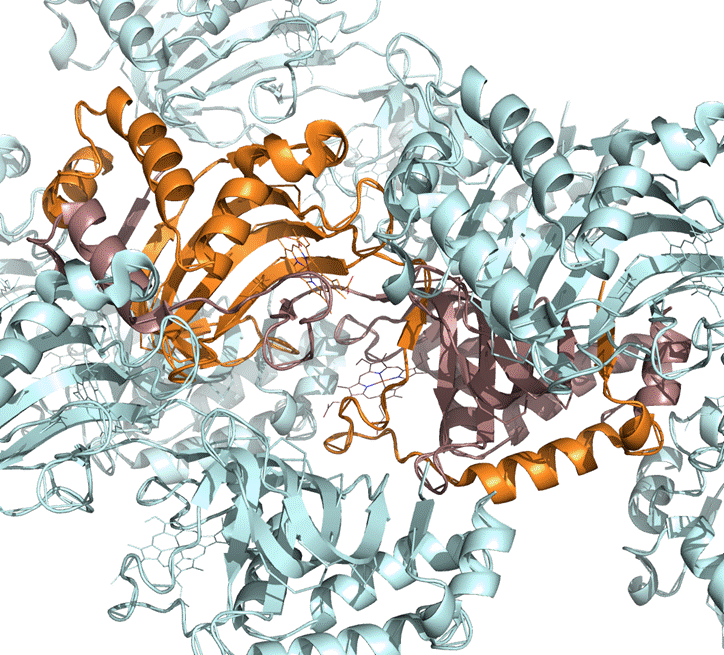
Тут молекулы гема взаимодействуют с тремя цепями.
Скорее всего данное различие обуславливается ошибками кристаллизации.
Структура 2cn4 была получена на 7 лет позже (в 2006ом), так что стоит предполагать, что более поздняя структура – более достоверная, к тому же укладка белка, взаимодействие между цепями димера и место связывания молекул гема выглядят гораздо правдоподобней в 2cn4.
Подтверждение этой версии можно найти в литературе, в статье Mirjam Czjzek и др. под названием “The Crystal Structure of the Secreted Dimeric Form of the Hemophore HasA Reveals a Domain Swappingwith an Exchanged Heme Ligand”, напечатанной в 2007 (т. е. через год после второй расшифровки и публикации структуры гемафора HasA). В ней правильность структуры 2cn4 доказывается методом ЯМР.
