
Партнерка на США и Канаду по недвижимости, выплаты в крипто
- 30% recurring commission
- Выплаты в USDT
- Вывод каждую неделю
- Комиссия до 5 лет за каждого referral
Аутосомно-рецесивні форми несиндромальної приглухуватості
Фіщук Л. Є.
ДУ «Інститут генетичної та регенеративної медицини АМН України»
Резюме. У статті представлено сучасні дані вітчизняної та зарубіжної літератури про аутосомно-рецесивні форми несиндромальної приглухуватості. Більше 50% аутосомно-рецесивних форм приглухуватості пов’язано з мутаціями в гені CJB2, який кодує коннексін 26 – мембранний білок. Мутація 35delG є найбільш частою і складає близько 70% всіх мутацій в гені CJB2 у європейській популяції.
Ключові слова: сенсоневральна приглухуватість, CJB2, коннексін 26, 35delG.
Cенсоневральна приглухуватість (далі – СНП) – це приглухуватість, обумовлена ураженням внутрішнього вуха, нейронів слухового шляху, слухової зони кори головного мозку, що супроводжує цілий ряд захворювань [1]. За різними даними, від цього захворювання потерпає від 6 до 10% населення України. Уражує хвороба осіб практично всіх вікових категорій – від дітей до літніх людей. Проблема порушення слуху у новонароджених дуже важлива як з медичної так і з соціальної точки зору. Зниження слуху у дитини, на відміну від дорослого, призводить до відхилення у мовному розвитку, формуванні інтелекту та особистості в цілому, особливо при виникненні приглухуватості та глухоти у новонароджених та дітей раннього долінгвального віку. За даними Всесвітньої організації охорони здоров’я, на 1000 новонароджених з нормальним слухом припадає одна дитина з вираженим ступенем приглухуватості, а порушення слуху легкого та середнього ступеню проявів присутнє у 1-2% новонароджених [2]. За даними вчених різних країн (США, Канади, Мексики, Англії, Данії, Японії) частота вродженої приглухуватості становить від 0,8 до 15,5 на 1000 новонароджених [3]. Крім цього, протягом перших 3 років життя приглухуватість проявляється ще у 2-3 дітей з цієї тисячі.
Раннє виявлення вродженої приглухуватості дуже важливе, так як час початку лікування є визначальним чинником ефективності реабілітації дітей з приглухуватістю. Своєчасний і правильний діагноз надає можливість більш успішної інтеграції дитини у мовне середовище [3].
Не меншою проблемою є слухова дисфункція у дорослих, яка формується у 5-14% людей між 45 та 64 роками, у 30% – після 65 років і у 50% у віці 80 років [4]. Приглухуватість, що розвивається у дорослих, має багатофакторну етіологію, серед якої важливе місце відводиться генетичним чинникам. Так, наприклад, доведено, що прийом деяких антибіотиків аміноглікозидного ряду (канаміцин, стрептоміцин та ін.) при наявності мутації A1555G в гені 12S rRNA може призвести до втрату слуху [5].
Серед спадкових порушень слуху виділяють:
· синдромальні, при яких приглухуватість поєднується із діагностованими змінами в роботі інших органів або видимими фенотипічними проявами (синдром Ваарденбурга, синдром Альпорта, синдром Ушера та ін. – складають близько 30% всіх випадків спадкових порушень слуху);
· несиндромальні, коли зниження або втрата слуху є єдиним проявом захворювання і не супроводжується іншими ознаками або захворюваннями інших органів і систем ( з частотою близько 70%).
Генетичні форми порушення слуху можуть успадковуватись за аутосомно-домінантним, аутосомно-рецесивним, Х-зчепленим, мітохондіальним, змішаним типом. Серед генетично-обумовленої приглухуватості в цілому аутосомно-рецесивні варіанти є найбільш частими – вони складають більше 70% всієї спадкової несиндромальної патології слуху [6]. Аутосомно-рецесивні форми СНП, зазвичай, є найбільш важкими і майже виключно пов’язані з кохлеарними дефектами.
Більше 50% аутосомно-рецесивних форм приглухуватості пов’язано з мутаціями в гені CJB2 (gap junction protein, beta 2), що локалізований в області 13q11-q12 в позиції 665037 [7]. CJB2 кодує коннексін 26 (Сx26) – мембранний білок, який із своїх шести субодиниць формує напівканал. При сполученні двох напівканалів сусідніх клітин формується так званий щілинний контакт – канал, який забезпечує пасивну дифузію електролітів, вторинних месенджерів і метаболітів (малих молекул розміром до 1кДа) із цитоплазми однієї клітини в цитоплазму іншої, сусідньої [8].
Коннексін 26 виявлений у всіх органах і тканинах, але його зміни значною мірою відображаються тільки на органі слуху. Відсутність ураження інших органів пояснюють тим, що у внутрішньому вусі він не може бути заміщений на інший вид коннексіну, як це відбувається в багатьох тканинах. У внутрішньому вусі коннексін 26 експресується в судинній смужці (stria vascularis), базилярній мембрані (basement membrane), спіральному лімбі (spiral limbus ) і спіральному виступі (spiral prominence) завитку [9].
Функціонально коннексінові канали відіграють ключову роль в рециркуляції іонів калію в епітелії равлика. Висока концентрація калію в ендолімфі забезпечує механізми електричної трансдукції при потраплянні його всередину чутливих клітин. Повернення калію в ендолімфу равликого ходу проходить завдяки пасивній дифузій через коннексінові канали підтримуючих клітин у напрямі до судинної смужки равлика, яка завдяки спеціальним каналам секретує калій назад в ендолімфу [10]. Молекулярна патофізіологія порушення функції Сх26-каналу в результаті мутації гена CJB2 визначається різним ступенем пригнічення рециркуляції іонів калію. Обструкція коннексінового щілинного контакту між чутливими і підтримуючими клітинами для дренажу іонів калію, що надходить у волоскові клітини в процесі механо-електричної трансдукції, швидко призводить до зниження його зворотнього надходження в судинну смужку і, як наслідок, до зменшення сецернування в ендолімфу. В результаті пригнічення електролітного гомеостазу ендолімфи знижується електричний потенціал на межі ретикулярної пластини. Результатом є пригнічення рецепторного потенціалу чутливих клітин і збудження аферентних нервових волокон. Стан різко погіршує патологічна гіперкалійгістія спірального органу, що призводить до калієвої інтоксикації нейроепітелію. Наслідком останньої є неконтрольоване зростання концентрації вільних іонів кальцію всередині чутливої клітини внаслідок мобілізації із депо ретикулуму і стимуляції входу через базилярну мембрану. Надлишкові вільні іони кальцію в цитоплазмі чутливої клітини є потужним проапоптичним стимулом [9]. Таким чином, порушення рециркуляції іонів калію призводить до незворотньої дегенерації чутливого епітелію та стійкої втрати слухової функції.
Причиною приглухуватості, асоційованої з коннексіном 26, можуть бути різноманітні мутації гена CJB2, яких на сьогоднішній день відомо більше 90 [11]. Найбільш частою, і відповідно, більш вивченою є мутація 35delG або 30delG (rs) – делеція одного з шести нуклеотидів гуанозинів (G) між положеннями 30 і 35 включно (рис.1) [12]. Мутація 35delG призводить до зсуву рамки зчитування, що веде до утворення стоп-кодону у 38 нуклеотиді, наслідком чого є передчасна зупинка синтезу білка Cx26.
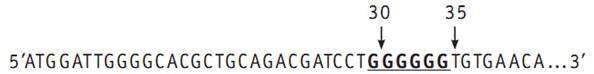
Рис. 1 Фрагмент кодуючої ділянки гена CJB2 з полі-G послідовністю в позиціях 30-35.
На частку цієї мутації припадає близько 70% всіх мутацій в гені CJB2 у європейській популяції [13], а загальний рівень носіїв складає 2,3-4% і залежить від регіону [14]. Середня частота мутації 35delG для європейської, американської, азіатської та африканської популяції складає, відповідно, 1,89, 1,52, 0,64 і 0,64 [15]. Вважають, що однонуклеотидна делеція 35delG є причиною 10% всіх ранніх дитячих слухових порушень і 20% всіх дитячих спадкових слухових порушень [16].
Завдяки тому, що мутація 35delG переважно поширена серед білого населення Європи, вона є основним претендентом для дослідження при установленні спадкових причин порушення слуху на території України. З найбільш поширених мутацій Cx26, що відповідають за розвиток несиндромальної рецесивної глухоти у небілого населення відомі:
· 167delT у євреїв-ашкеназі – частота у хворих зі спадковою глухотою становить біля 4%, а частота гетерозиготного носійства цієї мутації серед індивідів, що нормально чують, в популяції євреїв-ашкеназі становить 0,73% [17];
· 235delC – у азіатів – у хворих на глухоту японців мутація 235delC виявляється з частотою 70%, тоді як частота гетерозиготного носійства серед здорового населення складає 3,2% [18].
Клінічні прояви наявності мутацій в гені CJB2 зведені в таблиці 1 [19].
Таблиця 1
Клінічна характеристика носіїв мутацій в гені CJB2 в гомозиготному стані
Клінічна характеристика | Гомозиготи за мутаціями в гені CJB2 (35delG, 235delC та інші) | |
Характерні ознаки | Можливі ознаки | |
Належність до синдромальних форм | Несиндромальна СНП | Вроджені вади розвитку, інша супутня патологія |
Тип успадкування | Аутосомно-рецесивний | - |
Тип приглухуватості | Двостороння СНП | Виражена асиметрія двостороннього ураження |
Ступінь важкості | Глухота, важкі і помірно важкі ураження слуху | Середній ступінь |
Вік початку | Вроджений характер, виявлення в долінгвальний період | Виявлення до 5 років, у рідких випадках не пізніше 6 років |
Мовні навички | Затримка мовного розвитку різного ступеня вираженності | Нормальний мовний розвиток можливий при менш важких формах |
Прогресування | Ні, стабільний характер | Прогресування невстановленого генезу |
Крім Сх26 існують інші протеїни цієї родини, наприклад, коннексін-30, -31, -43, мутації в генах яких також формують картину аутосомно-рецесивної несиндромальної втрати слуху [20].
Виявлення у дітей в період новонародженності мутацій, пов’язаних з виникненням сенсоневральної приглухуватості, дозволить раніше встановити діагноз, що важливе для своєчасного початку лікування. Рядом досліджень показано, що найбільш ефективним шляхом реабілітації дітей з мутацією 35delG в гені CJB2 при тяжкому ступені приглухуватості є використання кохлеарної імплантації [21]. Таким чином, результати генетичного аналізу можуть бути використані при виборі тактики лікування. При медико-генетичному консультуванні сімей з випадками СНП батьки зможуть вчасно отримати інформацію про ризик виникнення повторного випадку захворювання в родині та можливість застосування пренатальної діагностики при наступних вагітностях.
Література
1.Міністерство охорони здоров’я. Наказ № 000 від 21.04.2005 «Про затвердження Протоколів надання медичної допомоги дітям за спеціальністю «дитяча отоларингологія»».
2.Шилова слуха у новорожденных детей / Н. А. Шилова, Н. В. Харламова, Т. В. Чаша, Н. Ю. Куликова, Е. В. Толкачева // Здоровье ребенка [Електронний ресурс]. – 2010. – №6(27). – Режим доступу: http://pediatric. /archive/issue-15373/article-15405.
3. Нарушение слуха у новорожденных детей / // Лечащий врач [Електронний ресурс]. – 2005. – №1. – Режим доступу: http://www. *****/2005/01/4531983.
4.Morton N.E. Genetic epidemiology of hearing impairment / N.E. Morton // Annals of the New York Academy of Science. – 1991. – Vol.630. – С.16-31.
5.Джемилева генов 12S rRNA и tRNASer(UCN) мтДНК у больных несиндромальной сенсоневральной тугоухостью/глухотой из различных регионов России / , , , // Генетика. – 2009. – Т.45, №7. – С. 982-991.
6.Smith R. J.H. Deafness and Hereditary Hearing Loss Overview / R. J.H. Smith, M. S. Hildebrand, G. Van Camp // GeneReviews [Електронний ресурс]. – 2010. – Режим доступу: http://www. ncbi. nlm. nih. gov/books/NBK1434
7.GJB2 gap junction protein, beta 2, 26kDa [ Homo sapiens ]: updated on 20.03.2011 [Електронний ресурс] / Bethesda: National Institute of Health, USA. – 2011. – Режим доступу: http://www. ncbi. nlm. nih. gov/gene? term=CJB2
8. Физиология межклеточной коммуникации: учеб. пособие / . – Минск: БГУ, 2008. – С.9-10.
9.Lefebvre P.P. Connexins, hearing and deafness: clinical aspects of mutations in the connexin 26 gene / P.P. Lefebvre, T.R. Van De Water // Brain Research Reviews. – 2000. – Vol.32. – P.159-162.
10. Генетический скрининг среди детей с врожденной и ранней детской тугоухостью / Т. Г. Маркова, Н. В. Некрасова, И. А. Шагина, // Вестник оториноларингологии. – 2006. – №4. – С.9-14.
11. Даниленко природа сенсоневральной тугоухости у жителів Беларуси / , , -Смоляк, , О. Г. Давыденко // Клинико-лабораторный консилиум. – 2010. – №2-3(33-34). – С.42-44.
12. Single Nucleotide Polymorphism Database: Entrez SNP: updated on 23.03.2011 [Електронний ресурс] / – Bethesda : National Institute of Health, USA. – 2011. – Режим доступу: http://www. ncbi. nlm. nih. gov/snp/
13. Lucotte G. The 35delG mutation in the connexin 26 gene (GJB2) associated with congenital deafness: European carrier frequencies and evidence for its origin in ancient Greece / G. Lucotte, F. Diéterlen // Genetic Testing. – 2005. – Vol.9. – P.20-25
14. Oliveira C. A. Allelic Frequencies of The 35delG Mutation of The GJB2 Gene in Different Brazilian Regions / C. A. Oliveira, C. J. Pimpinati, F. Alexandrino, L. A. Magna, A. T. Maciel-Guerra, E. L. Sartorato // Genetic Testing. – 2007. – Vol.11, №1. – Р.1-3.
15. Mahdieh N. Statistical study of 35delG mutation of GJB2 gene: a meta-analysis of carrier frequency / N. Mahdieh, B. Rabbani // International Journal of Audiology. – 2009. – Vol.48, №6. – Р. 363-370.
16. Журавский генетика несиндромных наследственных форм сенсоневральной тугоухости (Обзор литературы) / , Т. Сетхияасиилан, , // Российская оториноларингология. – 2004. – Т.7, №6. – С.164-177.
17. Morell R. J. Mutations in the connexin 26 gene (GJB2) among Ashkenazi Jews with nonsyndromic recessive deafness / R. J. Morell, H. J. Kim, L. J. Hood, L. Goforth, K. Friderici, R. Fisher, G. Van Camp, C. I. Berlin, C. Oddoux, H. Ostrer, B. Keats, T. B. Friedman // The New England journal of medicine. – 1998. – Vol.339, №21. – P..
18. Kudo T. Novel mutations in the connexin 26 gene (GJB2) responsible for childhood deafness in the Japanese population / T. Kudo, K. Ikeda, S. Kure, Y. Matsubara, T. Oshima, K. Watanabe, T. Kawase, K. Narisawa, T. Takasaka // American Journal of Medical Genetics. – 2000. – Vol.90, № 2. – Р. 141-145.
19. Клиника нарушений слуха, обусловленных изменениями в гене коннексина 26 / , , // Вестник оториноларингологии. – 2008. – №2. – С.4-9.
20. Ballana E. Connexins and deafness Homepage: updated on 27.03.2011 [Електронний ресурс] / E. Ballana, M. Ventayol, R. Rabionet, P. Gasparini, X. Estivill. – 2011. – Режим доступу: http://www. crg. es/deafness
21. Староха имплантация – перспективное направление слухопротезирования / , // Бюллетень сибирской медицины. – 2004. – № 4. – С.34-38.
Аутосомно-рецессивные формы несиндромальной тугоухости
Резюме. В статье представлены современные данные отечественной и зарубежной литературы про аутосомно-рецессивные формы несиндромной тугоухости. Более 50% аутосомно-рецессивных форм тугоухости связано с мутациями в гене CJB2, который кодирует коннексин 26 – мембранный белок. Мутация 35delG является частой и составляет около 70% всех мутаций в гене CJB2 в европейской популяции.
Ключевые слова: сенсоневральная тугоухость, CJB2, коннексин 26, 35delG.
An autosomal recessive nonsyndromic form of sensorineural hearing loss
Fischuk L. Е.
Summary. This article presents current data of domestic and foreign literature on autosomal recessive nonsyndromic forms of hearing loss. Over 50% of autosomal recessive forms of hearing loss associated with mutations in the gene CJB2, which encodes connexin 26 – membrane protein. 35delG is the most common mutation and is about 70% of all mutations in the gene CJB2 in European populations.
Key words: sensorineural hearing loss, CJB2, connexin 26, 35delG.
