

Партнерка на США и Канаду по недвижимости, выплаты в крипто
- 30% recurring commission
- Выплаты в USDT
- Вывод каждую неделю
- Комиссия до 5 лет за каждого referral
Федеральное агентство по образованию
Московская государственная академия
тонкой химической технологии им.
Кафедра коллоидной химии
Кафедра информационных технологий
,
Адсорбция на границе раздела «твердое тело – раствор»
Учебное пособие
Москва 2005 год
УДК 541.183
ББК 24.6
Рецензент:
Д. х.н., профессор
,
Адсорбция на границе раздела «твердое тело – раствор»
Учебное пособие
М.: МИТХТ им. , 2005.
Утверждено Библиотечно-издательской комиссией
МИТХТ им в качестве учебного пособия
Поз.
Данное учебное пособие соответствует программе курсов «Коллоидная химия» и «Поверхностные явления и дисперсные системы» для направления бакалавриата и магистратуры по направлению 550800 «Химическая технология и биотехнология» очной формы обучения. В нем подробно излагаются закономерности адсорбции индивидуальных жидкостей, растворов, ионов, поверхностно-активных веществ и высокомолекулярных соединений на твердом теле.
©МИТХТ им. , 2005 г.
1.Введение.. 4
2. Адсорбент и адсорбтив.. 4
2.1. Адсорбент.. 5
2.2. Адсорбтив.. 9
3. Природа адсорбционных сил.. 21
4. Механизм адсорбции.. 26
5. Теория адсорбции из бинарных растворов.. 30
6. Адсорбция низкомолекулярных несмешивающихся и смешивающихся жидких систем 34
7. Адсорбция поверхностно-активных веществ на твердых поверхностях.. 39
7.1. Адсорбция неионогенных ПАВ.. 40
7.2. Адсорбция ионогенных ПАВ.. 42
7.3. Пленки Ленгмюра-Блоджетт.. 50
8. Адсорбция электролитов (ионов) 53
9. Адсорбция полимеров на твердом теле. 64
10. Методы исследования адсорбции на границе раздела раствор - твердое тело.. 77
10.1. Термодинамические методы... 77
10.2. Методы, связанные со спектроскопией и рассеянием... 77
10.3. Гидродинамические методы... 78
10.4. Электро-химическне метода.. 78
10.5. Методы разъединяющего давления.. 79
11. Список литературы... 80
1.Введение
Адсорбцией называют процесс выравнивания химических потенциалов между поверхностью и объемом, приводящий к изменению концентрации компонента на поверхности.
Адсорбция из раствора на твердой поверхности процесс более сложный, чем адсорбция газов и индивидуальных жидкостей. Она лежит в основе важнейших явлений коллоидной химии, как получения, так и разрушения лиозолей.
Изучение адсорбции сжиженных газов, низкомолекулярных индивидуальных жидкостей на твердых поверхностях показало, в основном, применимость уравнений Генри, Бедеккера-Фрейндлиха, Ленгмюра и БЭТ. Однако, присутствие в системе, как минимум, третьего компонента во многом усложняет математическое описание и теоретическую трактовку адсорбции из смеси. Это связано, во-первых, с наличием взаимодействия между растворителем и растворенным веществом, во-вторых, значительной конкуренции за активные центры поверхности и, в-третьих, замедлением процесса диффузии растворенного вещества к поверхности из-за вязкости среда и созданием граничного слоя. Уплотненный граничный слой, состоящий из растворителя и растворенного вещества, препятствует обмену компонентов на поверхности и значительно замедляет установление равновесия в системе.
Явление адсорбции из растворов классифицируют по свойствам адсорбента на адсорбцию неэлектролитов (молекулярную) и электролитов (ионную).
2. Адсорбент и адсорбтив
Адсорбент - тело, на котором происходит концентрация вещества, может быть жидкостью, аморфным или твердым.
Адсорбтив - множество молекул, ионов с сольватными оболочками, отдельные макромолекулы или их агрегаты, коллоидные частицы, движущиеся во всех направлениях.
Рассмотрим поведение одной выбранной молекулы или агрегата. Сталкиваясь в объеме с себе подобными она меняет скорость и направление движения и может встретить на своем пути поверхность, например, стенку сосуда. В большинстве случаев молекула, ударившаяся о поверхность, на некоторое время остается на ней. Продолжительность пребывания зависит от множества факторов: от места на поверхности, природы поверхности и самой молекулы, специфики сил взаимодействия между поверхностью и молекулой и самими молекулами, кинетической энергии молекулы. При этом столкновении концентрация молекул на поверхности несколько отличается от объема.
Де Бур предложил расчет количества молекул газа, ударяющихся за одну секунду об 1 см2 поверхности при давлении 1 атм при комнатной температуре: для водорода - 11.1023 , для азота - 2,9.1023, для кислорода - 2,7.1023. Воздух с влажностью 10%, давление водяного пара порядка 1,75 мм рт. ст. способствует соударению 8,5.1020 молекул воды с 1 см2 поверхности в 1 секунду. А для покрытия мономолекулярным слоем 1 см2 поверхности достаточно 1015 молекул воды, то полное равновесие достигается за одну миллионную долю секунды. Следовательно, в растворе даже большие по размеру молекулы, агрегаты, ассоциаты, коллоидные частицы, фракталы, наночастицы способны за достаточно короткое время образовывать монослой, а динамическое равновесие зависит от множества факторов и зачастую не может быть описано единой теорией.
2.1. Адсорбент
Адсорбция молекул из жидкой фазы на твердом теле определяется интенсивным силовым полем, зависящим от состава и структуры. Поверхность всегда неровна (рис.1), и разные грани обладают различной адсорбционной способностью, являясь активными центрами адсорбции. Не все атомы на поверхности находятся в одинаковых условиях. Атомы на острых выступах и шероховатостях намного богаче энергией и обладают избыточной поверхностной энергией и подвижностью. Наряду с гранями, выступами большое значение имеют дефекты структуры реальных кристаллов, подробно изучаемых в курсе физики твердого тела (шероховатости, трещины, дислокации, грани, впадины, ребра, дефекты, выщербены, сколы).
|

Рис. 1. Шероховатость поверхности и активные центры
(по степени активности)
1>2>3>6>5>4
Поверхностное натяжение твердого тела в отличие от поверхностного натяжения жидкости неоднозначная величина. Это связано с тем, что при образовании новой поверхности атомы поверхностного слоя перегруппировываются и перемещаются в конечное равновесное положение в течение длительного времени. Образующаяся при этом грань кристалла принимает такую форму, чтобы суммарная свободная энергия кристалла данного объема была наименьшей. Кристаллы с ковалентными связями (например, алмаз) обладают поверхностной энергией плоскости (111) порядка 5650 мДж/м2, а для плоскости (100) эта величина около 9800 мДж/м2. Ионные кристаллы имеют значительно меньшие величины поверхностной энергии и несимметричность иона вблизи поверхности. Расчеты для хлористого натрия (NaCl) тех же граней (1мДж/м2, (1мДж/м2. Молекулярные кристаллы, к которым относится большинство твердых тел и все органические соединения, в том числе и полимеры, характеризуются еще меньшими значениями поверхностной энергии, от 18 до 80 мДж/м2.
Дефекты кристаллических решеток значительно сказываются на свойствах кристалла и поверхностной энергии. Наличие точечных дефектов в результате образования избыточных, внедренных атомов или ионов, а также дефекты, вызванные недостатком атомов в решетке - вакансии, приводят к появлению дислокации (совокупности дефектов). Дислокации образуются на поверхности в виде ступенек и находятся в состоянии высокого поверхностного напряжения. Химический потенциал такой дислокации настолько велик, что молекулы вещества передвигаются на поверхности, оставляя пространство в виде полости, являясь центром адсорбции. В зависимости от природы твердого тела число линий дислокаций, приходящихся на единицу поверхности кристалла, может изменяться в широких пределах от 106 до 1016 дислокаций на м2.
Выступы, ребра, грани и вершины кристаллических решеток обладают значительным числом ненасыщенных химических связей, большой суммарной поверхностной энергией, являются центрами хемосорбции, возникающей в результате близкодействующих электростатических сил притяжения. А дальнодействующие межмолекулярные силы, наоборот, увеличиваются по мере возрастания числа окружения соседей. Таким образом, центры физической адсорбции, в основном, находятся в трещинах, зазорах, впадинах. Следовательно, микротопография поверхности, наряду с химической природой и структурой, позволяет с большим приближением рассчитать средние значения поверхностной энергии с применением современных методов исследования поверхности, таких как:
1. Микроскопия:
a) оптическая - огранка поверхности и развитие дислокации,
b) электронная - поверхность с разрешением до 10Å,
c) сканирующая с рентгеновским анализатором – поверхность с разрешением до 100 Å и анализом состава,
d) эллипсометрия - измерение толщины пленок по изменению поляризации светового пучка для пленок толщиной до 1 Å,
2. Дифракция и рассеяние электронов и ионов:
a) дифракция медленных (ДМЭ-I-5ООЭВ) и быстрых (ДБЭ-18-25 КЭВ) электронов,
b) автоэлектронная (30-50 Å) и автоионная (0-0,5 Å) эмиссия,
c) рассеяние ионов,
3. Спектроскопия:
a) инфракрасная (ИКС) и комбинированное рассеяние (КРС), в том числе НПВО и МНПВО,
b) электронноспиновый (ЭПР) и ядерно-магнитный резонанс (ЯМР),
c) ОЖЕ-спектроскопия,
d) фотоэлектронная (ЭСХА),
e) Масс-спектроскопия вторичных электронов (МСВЭ),
4. Поверхностная проводимость (для полупроводников) и другие.
Наличие дефектов на поверхности, а также в объеме приводят к увеличению истинного значения удельной поверхности твердого тела. Гладкие (идеальные) поверхности встречаются очень редко. К ним можно отнести стекла, полимеры, керамику и специально приготовленные поверхности. В основном твердые тела обладают значительной пористостью. К ним относятся уголь и торф, изделия из древесины, ткани, зерно, кожа, почва и грунты, губчатые изделия, пенопласты и многие другие. Пористые тела обладают различной адсорбционной способностью по отношению к раствору или растворителю, механической прочностью, удельной поверхностью, селективностью и другими специфическими свойствами. Наиболее широкое применение в технике имеют активированные угли, сажи, силикагели и алюмогели, цеолиты (алюмосиликат со строго регулярной кристаллической структурой), пористые стекла, мембраны, целлофан.
Согласно классификации по Дубинину с учетом размеров пор и механизма протекающих в них адсорбционных процессов пористые тела делятся на:
1. Макропористые - поры с радиусом более 100-200 нм и удельной поверхностью от 0,5 до 2,0 м2/г. В основном играют роль транспортных каналов и адсорбцией в них пренебрегают.
2. Переходнопористые - поры с радиусом от 2,0 до 100 нм и удельной поверхностью от 10 до 500 м2/г - силикагели, алюмогели, алюмосиликагели.
3. Микропористые - поры с радиусом от 0,5 до 2,0 нм и удельной поверхностью от 500 до 1000 м2/г - активированные угли, цеолиты.
4. Супермикропористые - размер пор меньше 0,5 нм - дефекты и дислокации кристаллических решеток.
Для характеристики пористых тел недостаточно знать только размер пор или капилляров, которые отличаются количеством на единицу объема, размером и длиной капилляра. Поэтому принято характеризовать пористость структуры тела величиной пористости - отношением объема пор (Vп) к общему объему тела (Vобщ).
![]() (1)
(1)
Однако классификация тел не учитывает возможность одновременного присутствия открытых, закрытых и тупиковых пор с различной длиной капилляра. Закрытые, внутренние поры, дефекты кристаллической решетки и дислокации внутри кристалла не могут быть определены обычными методами, а применение томографии не позволяет однозначно рассчитать пористость. Поэтому пористость складывается из трех составляющих:
![]() (2)
(2)
где Пз, По, Пт - объемы соответственно закрытых, открытых и тупиковых пор в единице объема твердого тела.
При адсорбции следует учитывать не только количество тех или других пор, но и кривизну капилляра, сужение и расширение, а также распределение пор по размерам (рис.2).
|

Рис. 2. Примеры пор в твердом теле
1) тупиковая, 2) открытая, 3) закрытая, 4) открытая с утолщением внутри,
5) открытая с утоньшением внутри, 6) бутылочная, 7) гребенчатая,
8) поверхностная.
Кроме плоских поверхностей в большинстве случаев в процессе адсорбции участвуют нити, имеющие незначительную удельную поверхность и, главным образом, высокодисперсные твердые частицы. По размеру частиц дисперсные системы делятся на:
1) высокодисперсные - 10-7-10-9м (наночастицы),
2) микрогетерогенные - 10-5-10-7м,
3) грубодисперсные-10-3-10-5м (макрогетерогенные).
Тонкодисперсные твердые частицы получаются как измельчением грубодисперсных систем - диспергирование, так и конденсацией. В последнем случае свободнодисперсные системы: порошки или суспензии в большинстве случаев обладают заданным комплексом свойств - состав, удельная поверхность, форма и размер частиц, текучесть, насыпная плотность и другие. Введение зародышей кристаллизации позволяет получить системы с заданной полидисперсностью.
Дисперсность частиц определяет адсорбционную способность твердого тела и хорошо иллюстрирует влияние энергии ребер дробленого хлорида натрия при условии, что кубическая решетка при дроблении не искажается (табл. 1).
Таблица 1.
Зависимость удельной поверхностной энергии от размера частиц
хлорида натрия.
Длина ребра, м | Суммарная площадь, *104, м2 | Суммарная длина ребер, *102, м | Поверхностная энергия, мДж/кг | Энергия ребер, мДж/кг |
0,77*10-2 | 3,6 | 9,3 | 540*10-3 | 2,8*10-2 |
0,1*10-2 10-3 | 28 | 550 | 4,2 | 1,7*10-1 |
0,01 10-4 | 280 | 5,5*104 | 42 | 1,7*10-4 |
0,001 10-5 | 2,8*103 | 5,5*106 | 4,2*102 | 1,7*10-2 |
0,0001 10-6 (1мкм) | 2,8*104 | 5,5*108 | 4,2*109 | 1,7 |
0,000001 1Å) | 2,8*106 | 5,5*1012 | 4,2*1011 | 1,7*104 |
2.2. Адсорбтив
Адсорбтив, в отличие от твердого тела, состоит либо из отдельных молекул, либо смесей молекул. Концентрационная конформация и конфигурация молекулярных образований во многом зависят от взаимодействия между растворителем и растворенным веществом. Отдельными представителями являются растворы электролитов, хотя ионогенные ПАВ, мицеллообразующие системы, значительно отличаются от неорганических электролитов.
Растворители, широко применяемые в промышленности, делятся на неорганические и органические жидкости. К неорганическим, прежде всего, относится вода, кислоты и щелочи, сжиженные газы Н2, О2, N2, NH3, SO2, CO2 и другие. Особенно широко представлены синтетические органические растворители, отличающиеся природой функциональных групп, их количеством и молекулярной массой.
Необходимость классификаций широкого спектра растворителей привела к созданию разнообразия по подразделению их по тому или иному признаку.
1. По физическим свойствам
1) По диэлектрической проницаемости ( ε ):
a) с низкой (1,9-12) - гептан, гексан, фторуглероды...
b) со средней (12-50) - органические кислоты, спирты,
c) с высокой (> 50) - вода, глицерин, неорганические кислоты, основания.
2) По дипольному моменту
3) По вязкости
a) низковязкие (< 2 спз), Па. с. 10-3
b) средневязкие (2-10 спз), Па. с. 10-3
c) высоковязкие (> 10 спз), Па. с. 10-3
4) По температуре кипения
a) низкокипящие (< 100°С),
b) среднекипящие (100-150°С),
c) высококипящие (> 150°С).
5) По их способности к испарению (за 1 принято испарение этилового эфира при 20°С и относительной влажности 65- 70%)
a) легколетучие (< 10),
b) средне летучие (10-35),
c) труднолетучие (> 35).
2. По химическим свойствам.
Помимо известной классификации - кислоты, спирты, углеводороды, галогенамиды и т. д., и по кислотно-основному взаимодействию - протонногенные (кислые), протоннофильные (основные) и амфипротонные. Наиболее применима и разработана классификация растворителей по способности к образованию Н-связи.
I класс: жидкости, способные к образованию объемной трехмерной сетки Н-связи (вода, муравьиная кислота, гликоли) и имеют значительные величины ε и хорошо растворяются друг в друге.
II класс: жидкости, в которых возникают двухмерные сетки Н-связи (фенолы, одноатомные спирты и кислоты) - содержание одну ОН - группу.
III класс: жидкости, имеющие в составе молекулы атомы азота, кислорода, серы, фтора и другие, которые способны образовывать Н - связи с протоном партнера (эфиры, амины, кетоны и другие органические соединения.
IV класс: жидкости, молекулы которых имеют атом водорода, способный к участию в Н - связи, но не имеют атомов, которые могли бы быть акцепторами протона (хлороформ, дихлорэтан).
V класс: жидкости, молекулы которых не способны к образованию Н - связи ни в качестве доноров, ни в качестве акцепторов (четыреххлористый углерод).
Наиболее часто встречаемая жидкость со стандартными свойствами - вода, особенности свойств которой определены, прежде всего, строением ее молекул. Молекула воды имеет четыре полюса зарядов, расположенных в вершинах тетраэдра с расстоянием положительных зарядов до центра атома кислорода порядка 0,99 Å (рис. 3).
|
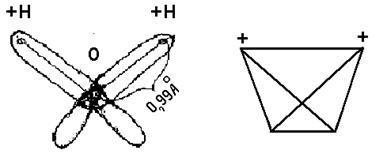
Рис. 3. Структура молекулы воды
Ажурность структуры воды объясняется в значительной мере образованием водородных связей между молекулами и определяет особенности диффузии ее молекул. В этом случае активированный переход отдельных молекул не требует "дырок", так как они имеются в самой структуре воды.
Изучение самодиффузии молекул воды и ионов в разбавленных растворах солей показало, что ионы в растворе не только гидростатируются, но и, главным образом, изменяют структуру самих молекул воды. Коэффициент самодиффузии воды в растворах солей значительно выше, чем в чистой воде, что связано с образованием гидратной оболочки и разрушением структуры прилегающих слоев воды. Происходит, с одной стороны, как бы "замораживание" молекул воды, а с другой, как бы плавление прилегающих слоев воды.
Проведенные исследования показали, что ион лития Li+ (r=0,68Å), обладающий высокой поляризуемостью, способен значительно упорядочить структуру воды, включенную в гидратную оболочку, по сравнению со свободной "несвязанной" водой. Ион цезия Cs+ (r=1,65Å) незначительно изменяет структуру воды в гидратной оболочке, но зато в сильной степени изменяет структуру воды в прилегающих к гидратированному иону слоях воды. Следовательно, из-за нарушения структуры воды вокруг иона вязкость чистой воды значительно падает, поэтому подвижность ионов Cs+ становится больше по сравнению с подвижностью ионов Li+ вместе с сольватной (гидратной) оболочкой. При этом самодиффузия чистой воды в разбавленных растворах Cs+ выше, чем в растворах Li+.
Гидратацию рассматривают не как связывание ионами того или иного числа молекул воды, а как влияние ионов на трансляционное движение ближайших молекул воды. Эксперимент показал, что некоторые ионы уменьшают подвижность ближайших молекул воды, а около других подвижность молекул воды значительно возрастает по сравнению с чистой водой, что определяет отрицательную гидратацию. Расчет показал, что полная энергия взаимодействия иона с молекулами воды необычайно велика и намного больше взаимодействия между молекулами воды друг с другом, и процесс обмена с ближайшими молекулами воды не связан с энергией удаления воды из сольватной оболочки. При этом частота обмена (коэффициент диффузии) определяется потенциальным барьером, отделяющим молекулы, связанные гидратной оболочкой, от молекул, находящихся в свободном состоянии. Следовательно, если обмен затруднен, то гидратация велика и, наоборот, по мере возрастания частоты обмена гидратация ослабляется.
Однако концентрированные растворы ионов электролита характеризуются структурами близкими к структурам соответствующих кристаллогидратов, а координационное число ионов соответствует координационному числу кристаллогидратов. Таким образом, наблюдается переход от структуры чистой воды к структуре кристаллогидрата по мере повышения концентрации иона в растворе. Существование обеих структур при повышенных концентрациях и низких температурах приводит к образованию "квазиэвтектических" растворов,
Льюис (1905г.) для характеристики поведения реальных систем предложил понятие активности - концентрация, которую имела бы исследуемая жидкая система, если бы она обладала свойствами идеальной системы.
![]() (3)
(3)
где а - активность системы, с - концентрация, γ - множитель (коэффициент активности).
Так как работа идеальной системы (Аид) в связи с изменением концентрации от с1 до с2 может быть рассчитана по уравнению
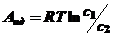 (4)
(4)
то для расчета работы реальной системы (А реал) это уравнение приобретает следующий вид
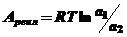 (5)
(5)
Химический потенциал μ вещества в идеальной и реальной смешанной фазе описываются следующими уравнениями
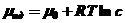 (6)
(6)
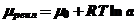 (7)
(7)
Из этого следует, что уравнение, ранее пригодное для идеальных систем, становится пригодным и для реальных систем. "Активность" как аргумент может рассматриваться как функция от термодинамических свойств растворов
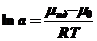 (8)
(8)
При соответствующих подстановках получим:
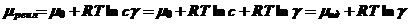 (9)
(9)
тогда коэффициент активности имеет вид:
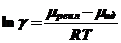 (10)
(10)
где lnγ - безразмерная величина, определяющая разность химических потенциалов в реальной и идеальной системах, и чем больше эта разница, тем больше энергии надо затратить, чтобы перевести реально существующую систему из реальной в идеальную (мера работы переноса одного моля растворенного вещества из реальной системы в идеальную).
Семенченко (1939 г.) вывел понятие средней потенциальной энергии одного моля (Un) с учетом межмолекулярного взаимодействия между растворенным веществом и растворителями, что осуществляется равенством
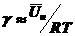 (11)
(11)
тогда
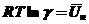 (12)
(12)
а коэффициент активности равен
![]() (13)
(13)
Наблюдающееся отличие коэффициента активности от концентрации вызваны изменением концентрации растворенного вещества в связи с образованием различных продуктов присоединения и сольватов, приводящих к уменьшению числа частиц в растворе, и значительному отличию энергии растворенных частиц, связанную с их взаимодействием между собой и с молекулами растворителя. Это особенно ярко проявляется у сильных электролитов, что определяется наличием значительного электростатического взаимодействия между ионами. В концентрированных растворах изменение коэффициента активности связывают с изменением свободной энергии из-за гидратации ионов.
|

Рис. 4. Зависимость средних коэффициентов активности от концентрации галогенидов щелочных металлов:
1 – LiF; 2 – LiCl; 3 – CsJ; 4 - CsCl
Как видно из рис.4 и 5, взаимодействие между растворенным веществом и средой (вода) во многом зависит от ионного радиуса и валентности катиона и аниона, а в неводных растворах еще накладывает определенное влияние диэлектрическая проницаемость среды.
Таким образом, рассматривая состояние растворов электролитов в реальных растворителях необходимо учитывать следующее:
1) равновесный состав и реакции взаимодействия в растворе, приводящие к этому составу;
2) характеристику сил, определяющих взаимодействие частиц растворенного вещества и молекул растворителя;
3) количественную характеристику зависимости между энергией сольватации ионов и молекул и энергией их взаимодействия в растворах;
4) общие закономерности, позволяющие охарактеризовать свойства образующихся растворов на основании данных о свойствах присутствующих в растворах веществ.
|

Рис. 5. Зависимость средних коэффициентов активности от концентрации электролитов:
1 - MgCl2; 2 - СаCl2; 3 - BaCl2; 4 - Li2SO4; 5 - Na2SO4, K2SO4.
Адсорбция между твердым телом и растворителем подчиняется известным законам и описывается элементарными уравнениями для двухфазных систем, поэтому в нашем случае не рассматривается.
Другими представителями, находящимися в виде молекул в водной среде и одновременно растворяющих молекулы воды, являются простейшие органические спирты и кислоты, которые изучены различными исследователями и представляют собой типичные модельные системы. Взаимные смеси различных органических растворителей сильновязкие системы и сложны для описания.
Существенным вопросом является ориентация молекул на поверхности раздела "жидкость - жидкость" или "жидкость - газ", зависящая от интенсивности силового поля, которая в свою очередь зависит от полярности и особенностей структуры жидкости. На границе раздела фаз молекулы ориентируются таким образом, чтобы обеспечить постепенный переход из одной фазы в другую, и чтобы энергия их взаимодействия была максимальной. Рассмотрим ориентацию молекул этанола на поверхности воды (рис. 6).
Согласно Ленгмюру (1924 г.), поверхностная свободная энергия молекулы аддитивно складывается из локальной свободной энергии этанола и воды. В первом случае поверхность покрыта гидроксильными группами, поверхностная энергия которых составляет порядка 190 мДж/м2. Во втором случае поверхностная энергия равняется поверхностной энергии углеводородов около 50 мДж/м2. Разность между представленными величинами составляет 140 мДж/м2 или 3.10-16 Дж/молекулу. Это убедительно доказывает преимущественную ориентацию органических молекул по типу 2 (рис. 6),
![]()
 ОН Н
ОН Н
Г Н С Н 



 Г Н С Н
Г Н С Н
![]()
![]()
![]()
![]()


Ж Н С Н 

 Ж Н С Н
Ж Н С Н
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
Н ОН
1 2
Рис. 6. Расположение молекулы этилового спирта на поверхности воды.
Это хорошо согласуется с величинами поверхностного натяжения большинства органических жидкостей и расчетов работ когезии и адгезии и описываются уравнением Гиббса.
Г2 = - (dσ/dμ2)T, (14)
где Г2 - избыток компонента 2 (этанол) в поверхностном слое площадью 1 м2 по сравнению с числом молей компонента в объеме раствора, содержащем такое же число молей растворителя, что и поверхностный слой (рис. 6).

Рис. 7. Изменение поверхностных избытков этанола в зависимости от состава раствора.
Таким образом, наличие дифильных молекул спирта приводит к неаддитивному распределению молекул этанола и воды друг в друге. Чем больше длина углеводородного радикала, тем хуже растворимость, взаимодействие между молекулами, тем интенсивнее расслаивается система. Это подтверждается известным правилом Траубе. Увеличение длины углеводородного радикала на СН2 - группу способствует увеличению поверхностной активности растворенного вещества в 3,2 раза. Это правомерно для гомологических рядов простейших органических соединений (С1 – С5) по отношению к воде и обратно для их растворов в органических растворителях. Эти вещества называются поверхностноактивными (ПАВ).
В случае, когда длина углеводородного радикала превышает 10-20 единиц групп - СН2 , содержит большое количество функциональных групп, не диссоциирует на ионы (неионогенные), принято говорить о коллоидных ПАВ. При малых концентрациях эти ПАВ находятся в растворителе в виде истинного раствора (отдельные молекулы) и к ним применимо правило Траубе, но, начиная с некоторой малой концентрации (критической концентрации мицеллообразования - ККМ) образуются в результате "гидрофобного взаимодействия" ассоциаты - мицеллы. Форма и размер мицелл с увеличением концентрации коллоидных ПАВ значительно отличается от сферической (рис. 8).
|
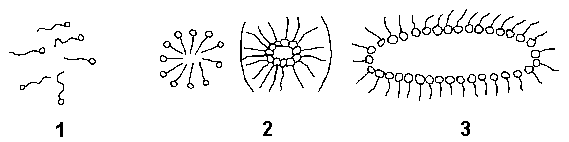
Рис. 8. Переход коллоидных ПАВ из молекулярного состояния (1) в глобулярное (2 – по Гертли) и цилиндрическое (3 – по Мак Бену).
Поэтому форма и размер мицелл коллоидных ПАВ должны привносить и значительные изменения в их адсорбционную способность.
ПАВ классифицируют следующим образом:
Поверхностно-активные вещества

 8 млн. т/в год
8 млн. т/в год
Ионогенные 70%(катионые, анионые, Неионогенные 30%
амфолитные, катионо-анионые)
1) Катионноактивные 60%
2) Анионноактивные >10%
3) Амфолитные
4) Катионо – анионоактивные 0,2%
Классификация:
1) Катионноактивные - растворимы в кислой среде
а) октадициламин - C18H37NH2
октадицилхлорамин
б) четырехзамещенный NH3 и пиридиновые основания, и их соли
алкилтриметиламмонийфторид RN (CH3)3CI, цетазол-

2) Анионактивные - растворимы в щелочной среде
а) карбоновые кислоты и их щелочи (мыла)
RCОOMe C17H35СОО-Na+ (олеат Nа)
б) алкиларилсульфонаты
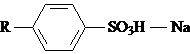
(70%) - очень дешевы
в) алилсульфонаты R – SO3Na (C15 – 20)
г) алкилсульфонаты (более дорогие) ROSO3H (C12 – 14)
д) алкенсульфонаты RНSО3 (C15 – 20)
Ценон Т – С17Н35 - С - О - N(CH3) - C2H4 - SO3Na
4) Амфолитные - диссоциируют в обеих средах, но по-разному Цетиламиноуксусная кислота C16H33NHCH2-СООН
катионоактивный | анионоактивный |
в кислой среде - NH2+ | в щелочной среде СОО- |
4) Катионо-анионоактивные
Дибутилнафталинсульфонат¯ (диметилфенилбензил]+ аммония

5) Неионогенные
а) октилфенолы ОП нонилфенолы NP - 8
Продукты полиоксиэтилирования к спиртам или фенолам
(RO)2 – P – (OC2H4)OH алкилортофосфорная кислота
б) плюроники - растворимы в воде
блоксополимеры окиси этилена (носитель гидрофобный) и окиси пропилена
(носитель гидрофобный) HO(С2Н4O)п - х(С3Н6О)м(С2Н4О)хН
в) маслорастворимые | растворимы в органике |
Эфиры жирных кислот и многоатомных спиртов |
|

дистеарат сульфат
Классификация ПАВ по молекулярному механизму действия (по Ребиндеру).
I группа: В системе вода - воздух не структурирующие, слабое пенообразование, слабые смачиватели - высшие спирты, амины, карбоновые кислоты.
II группа: В системе вода - масло не структурообразующие - диспергаторы, эмульгаторы, селективно смачивающие в системе твердое тело / жидкость.
III группа: структурообразователи, гидрофилизируют, стабилизируют, при малых концентрациях защищают коллоидные частицы от коагуляции, при больших концентрациях - пластификаторы - глюкозида, белки, полисахара.
IV группа: моющие вещества - обладают всеми свойствами трех предыдущих групп, а также обладают мицеллообразованием и солюбилизацией (включение растворенных молекул в объем мицеллы).
Следующими типичными представителями молекулярных растворов безусловно являются растворы высокомолекулярных соединений (ВМС). Обладая длинными и гибкими органическими макромолекулами с большой адсорбционной способностью, они хорошо растворяются в органических растворителях и образуют ассоциаты при высоких концентрациях. Однако, коренным отличием молекул ВМС от других вышеперечисленных растворенных веществ безусловно является чувствительность неионогенных молекул ВМС к природе растворителя.
Растворители по отношению к макромолекулам делятся на "хорошие", "плохие" и θ-растворители - условное обозначение, нашедшее подтверждение по соотношению параметров растворимости и описываемое следующим уравнением
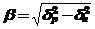 (15)
(15)
где - β - параметр совместности, (МДж/м3)1/2,
δр - параметр растворимости растворителя, (МДж/м3)1/2,
δп - параметр растворимости полимера,(МДж/м3)1/2.
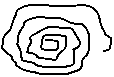
При β ≈ 0 - θ-растворитель, макромолекула представляет из себя гибкую цепочку (рис. 9, а),
при 0 < β < 0,2 - "хороший" растворитель, и макромолекула ВМС выглядит в виде эллипсоида вращения (рис. 9, б),
при 0,2 < β < 0,5 - "плохой" растворитель, и форма макромолекул наиболее выгодная - глобула - близкая к шарообразной (рис. 9, в).
 |
 |
а б в
Рис. 9. Форма макромолекул ВМС в различных растворителях:
а - в θ-растворителе, б - в "хорошем", в - в "плохом".
Совершенно по-другому ведут себя макромолекулы полимеров в концентрированных растворах. При этом концентрированные растворы делятся на: 1) разбавленные (1-3 мас.%), 2) умеренноконцентрированные (5-10 мас.%), 3) концентрированные (10-25 мас.%) и 4) высококонцентрированные (25-65 мас.%). С увеличением концентрации размер макромолекулярных образований проходит через максимум, но плотность упаковки индивидуального ассоциата увеличивается.
3. Природа адсорбционных сил
Взаимодействие молекул адсорбтива с твердой поверхностью может осуществляться либо за счет химических близкодействующих сил (хемосорбция), либо за счет физических, дальнодействующих сил притяжения (Ван-дер-Ваальса).
Наличие у поверхности активных центров, которые характеризуются высотой локализованных вблизи поверхности электронных энергетических уровней. В зависимости от расстояния между центром и адсорбированной молекулой может при сближении происходить отталкивание в результате индуцирования диполей взаимоориентированных вдоль электрического поля, либо сближение, приводящее к перекрытию электронных уровней молекул адсорбтива и адсорбата. примером может служить адсорбция воды на кремнезёме (рис. 10).
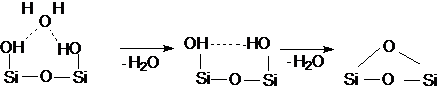
Физическая Химическая Дегидратация
адсорбция (1) адсорбция (2) адсорбата (3)
Рис. 10. Адсорбция воды на кремнеземе
Физически адсорбированная вода удаляется при температурах близких к 1ОО°С, а хемосорбированная требует повышения температуры до 300-400°С, дальнейшее увеличение температуры более 500°С приводит к деформации силиконовых групп и пасификации поверхности.
При хемосорбции идет изменение как свойств твердого тела (на поверхности), так и адсорбтива. При этом взаимодействие осуществляется электростатическими силами, описываемыми законом Кулона для точечных зарядов:
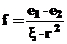 (16)
(16)
где f - сила: взаимодействия между ионами,
e1 и e2 - величина заряда иона,
ξ - диэлектрическая проницаемость,
r - расстояние между ионами.

Рис. 11. Кривые потенциальной энергии неактивированной (а) и активированной (б) хемосорбции
По потенциальной энергии образования прочного адсорбционного комплекса химические силы в основном можно расположить в следующий ряд:
ионные - с увеличением валентности возрастают, обратно пропорциональны квадрату расстояние (дальнодействующие), порядка Дж/моль
ковалентные или координационные - короткодействующие с небольшой энергией межмолекулярного взаимодействия (10-40 Дж/моль). В одном из наиболее успешных подходов хемосорбцию нередко рассматривают просто как реакцию, протекающую с образованием между твердым телом и адсорбтивом реакцию энергетически подобную образованию двухатомной молекулы.
При адсорбции ВМС, ПАВ или растворителей на твердых поверхностях наиболее существенный вклад оказывают физические силы (Ван-дер-Ваальсовые). Молекулярные физические силы взаимодействия получили свое название потому, что они обуславливают отклонение газов от идеального состояния, характеризующегося постоянной а в известном уравнении Ван-дер-Ваальса:
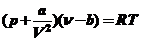 (17)
(17)
где a - поляризуемость молекул,
ν – частота колебаний атома.
К ван-дер-ваальсовым силам относятся:
1) Взаимодействие молекул с постоянным жестким диполем (ориентационный эффект) (силы Кеезома) описываемое уравнением
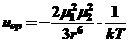 (18)
(18)
где uор - потенциальная энергия электростатического взаимодействия двух постоянных диполей,
μ1 и μ2- дипольные моменты обеих молекул,
r - расстояние между центрами диполей,
kT - энергия микроброуновского движения (постоянная Больцмана и абсолютная температура.
В зависимости от дипольного момента контактирующих молекул ориентационный эффект привносит до 50 % в общий вклад в межмолекулярное взаимодействие: чем меньше дипольный момент молекулы, тем меньше величина прочности связи.
Это взаимодействие осуществляется на близких расстояниях (1-5 А) и энергия ориентационных сил порядка 5-10 кДж/моль.
2) Взаимодействие молекул с постоянным диполем и молекул с индуцированным диполем (индукционный эффект) (силы Дебая) описываемое уравнением
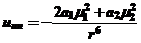 (19)
(19)
где UИН - потенциальная энергия взаимодействия двух различных молекул,
μ1 и μ2- дипольные моменты молекул,
а1 и а2 - поляризуемость молекул,
r - расстояние между диполями.
Поскольку температура не влияет на поляризуемость, индукционное взаимодействие в отличие от ориентационного не зависит от температуры. Энергия индукционного взаимодействия не превышает 5% от общего вклада ван-дер-ваальсовых сил и поэтому всегда имеет знак минус.
Расстояние, при котором возрастает индукционное взаимодействие, не превышает 1-2 А, а энергия связи мала и составляет не более 1-4 ккал/моль.
В ряде случаев молекулы, не имеющие постоянный или индукционный диполь, также способны взаимодействовать друг с другом за счет мгновенного дипольного момента. Даже между неполярными молекулами возникают силы межмолекулярного взаимодействия (дисперсионный эффект) (силы Лондона) и описываемые уравнением:
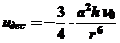 (20)
(20)
где uдес - потенциальная энергия взаимодействия двух неполярных атомов,
a - поляризуемость атома,
h - постоянная Планка,
ν0 - частота дисперсионного спектра колебаний атома,
r - расстояние между атомами.
Дисперсионные силы возникают из-за флуктуации электронной плотности в атоме адсорбата, которая индуцирует подобные флуктуации в атоме адсорбтива. Резонанс подобных флуктуации приводит к значительному снижению общей энергии системы в целом, приводящее к взаимному притяжению контактирующих неполярных молекул. Дисперсионное взаимодействие универсально и возникает у любых молекул, поэтому их доля от 50 до 100% в общем вкладе в прочность адсорбционного взаимодействия между молекулами и атомами. Дисперсионные силы не зависят от температуры и всегда вызывают только притяжение, поэтому имеют отрицательный знак.
Наиболее полно взаимодействие молекулы неполярного адсорбтива с атомом на поверхности неполярного твердого тела описывается уравнением Леннарда-Джонса
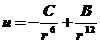 , (21)
, (21)
где u - общая энергия межмолекулярного взаимодействия двух молекул начиная с расстояния между ними равного нулю,
С - константа уравнения описывающая все три возможных варианта взаимодействия,
B - силы броуновского отталкивания, имеющие электростатическую природу и действующие на очень близких расстояниях.
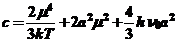 (22)
(22)
Дисперсионные силы являются аддитивными, суммирование их дает значительный эффект дальнодействия. Энергия притяжения молекул адсорбтива поверхностью адсорбата уменьшается с расстоянием гораздо медленнее, чем энергия притяжения молекул адсорбтива в системе. Из этого следует, что максимальное действие сил притяжения проявляется в углублениях, впадинах, трещинах твердого тела. Это можно объяснить тем, что силовое поле, образуемое взамопротивоположными стенками поры, щелей, углублений, накладывается друг на друга, и таким образом значительно усиливается.
Расстояние, на котором действуют дисперсионные силы, порядка 5-100 А, и энергия взаимодействия их превышает вышеперечисленные и составляет 10-30 кДж/моль.
При изучении адсорбции молекул на поверхности силикатного и алюмосиликатного типа адсорбционное взаимодействие объясняется возникновением водородных связей. Это объясняется тем, что у атомов 2-го периода таблицы (фтор, кислород, азот, а также у атомов серы и хлора) из-за малого радиуса ионов и заметному сродству к электрону появляется способность оттягивать электрон от соседствующего с ними атома водорода. Атом водорода в этом случае приобретает свойства протона Н+ и взаимодействует с электронами других элементов за счет во водородной связи.
Примером является образование водородной связи и айсберговая структура ассоциатов воды и льда

Подобные ассоциаты могут образовываться и у органических молекул
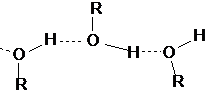
Большое количество молекулярных соединений, которые могут образовывать водородную связь принято выделять в особый класс ассоциирующих веществ. К ним относятся углевода, белки и ВМС,
"Гидрофобное взаимодействие". Термин "гидрофобное взаимодействие" прежде всего обозначает усиление связи между двумя частицами в присутствии растворителя, если взаимодействие типа растворитель - растворитель сильнее, чем взаимодействие растворитель - частица.
Примером может служить следующий эксперимент: 2 металлические пластинки, покрытые кремнеорганической жидкостью (гидрофобная) помещены в полярную жидкость - воду. При их соприкосновении они удерживаются друг около друга за счет "стягивающего" (гидрофобного) усилия, а те же пластинки, но не обработанные органической жидкостью, разделяются самопроизвольно под действием силы тяжести пластинки.
Выигрыш энергии, обусловленный сближением двух модифицированных металлических поверхностей, связан с действием ван-дер-ваальсовых(дальнодействующих) сил притяжения, а именно – дисперсионных согласно
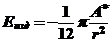 (23)
(23)
подробно описанное в предыдущих разделах.
С учетом наличия поверхности (1), гидрофобизирующэй жидкости (2) и среды (3) дисперсионное взаимодействие будет определяться константой Гамакера с учетом вклада всех сил
А*123 = А*12 – ( А*13 + А*23 + А*33 ) (24)
Следовательно дисперсионные силы А*123 значительно больше А*12 при условии, что А*33 > А*13 + A*23, что и наблюдается в нашем случае.
4. Механизм адсорбции
Наиболее предпочтительным подходом к изучению механизма адсорбции является исследование изотермы. Важнейшими характеристиками адсорбции безусловно являются
1) скорость адсорбции (константа скорости),
2) форма изотермы,
3) наличие плато на изотерме,
4) степень адсорбции растворителя,
5) тип адсорбции (мономолекулярная или полимолекулярная),
6) ориентация адсорбированных молекул,
7) влияние температуры на изотерму адсорбции,
8) природа взаимодействия между раствором и растворенным веществом,
адсорбатом и адсорбентом.
Различные формы рассматриваемых изотерм адсорбции представлены на рис. 11 по классификации Гильса.
Исходя из формы начального участка были выделены 4 характерные класса изотерм, а деление изотерм на отдельные типы внутри класса связано с последующим изменением их формы при более высоких концентрациях.
1. Класс S - начальный участок изотерм класса S выгнут относительно оси концентраций и при увеличении концентрации вещества получается точка перегиба, которая придает изотерме характерную S - образную форму.
2. Класс L (класс Ленгмюра) с наиболее общей и в начальной стадии изотермы этого класса характеризуются вогнутой линией относительно оси концентрации ( L1 в L2). По мере увеличения концентрации адсорбция достигает насыщения и приводит к образованию плато (L3) и протеканию процесса полимолекулярной адсорбции до достиже ния второго и последующих плато ( L4 ). L5 характерно для адсорбции ассоциатов ПАВ и красителей, но в чистом вида невозможен по термодинамическим причинам.
3. Класс Н - наблюдается при чрезвычайно сильной адсорбции при низких концентрациях. Обеспечено высоким сродством адсорбата и адсорбтива.
4. Класс C (постоянное распределение) наблюдается на микропористых телах.
Теоретический анализ различных типов изотерм адсорбции позволяет получить большую массу полезной информации о механизме адсорбции. Константа в уравнениях адсорбции (Ленгмюра, БЭТ и др.) в основном связана с энергией активации удаления растворенного вещества с поверхности. Если взаимодействие между молекулами растворителя и растворенного вещества мало, и мы можем им пренебречь, то энергия активации не зависит от степени заполнения поверхности, что приводит к изотермам типа L или H. Если сила взаимодействия между адсорбированными молекулами больше силы взаимодействия между растворенным веществом и твердым телом, энергия активации возрастает, а совместная адсорбция описывается, S-образной изотермой. В этом случае молекулы растворенного вещества стремятся расположиться на поверхности в виде цепей или кластеров. Подобному их расположению способствует сильная адсорбция растворителя и монофункциональный характер растворенного вещества.
 |
Рис. 12. Схема различных форм изотерм адсорбции по классификации Гильса
Теоретический анализ различных типов изотерм адсорбции позволяет получить большую массу полезной информации о механизме адсорбции. Константа в уравнениях адсорбции (Ленгмюра, БЭТ и др.) в основном связана с энергией активации удаления растворенного вещества с поверхности. Если взаимодействие между молекулами растворителя и растворенного вещества мало, и мы можем им пренебречь, то энергия активации не зависит от степени заполнения поверхности, что приводит к изотермам типа L или H. Если сила взаимодействия между адсорбированными молекулами больше силы взаимодействия между растворенным веществом и твердым телом, энергия активации возрастает, а совместная адсорбция описывается, S-образной изотермой. В этом случае молекулы растворенного вещества стремятся расположиться на поверхности в виде цепей или кластеров. Подобному их расположению способствует сильная адсорбция растворителя и монофункциональный характер растворенного вещества.
Примером различного типа изотерм адсорбции может послужить адсорбция фенола и резорцина на оксида алюминия (полярный) и вода (полярный). В первом случае процесс описывается изотермой S-типа, а во втором L-типа. Параллельная ориентация молекул резорцина приводит к занятию активных центров при малых концентрациях и поэтому приводит к изотермам типа L.
Красители и неионогенные ПАВ образуют в растворе агрегаты, и их адсорбция (циановые красители в водных растворах на галогенидах серебра) описывается изотермами S-типа.
Изотермы H - типа наблюдаются в тех случаях, когда адсорбция сопровождается образованием химического соединения (хемосорбция). Так адсорбция стеариновой кислоты из бензола на сильнополярных порошках металлов
Микропористые адсорбенты в основном характеризуются изотермой С - типа, так как число активных адсорбционных центров из-за их недоступности остается постоянным в широкой области концентраций. По мере заполнения молекулами адсорбтива одних активных центров появляются новые и доступная для адсорбции поверхность увеличивается пропорционально количеству адсорбированного из раствора вещества.
В основном, между адсорбцией из растворов твердых (при комнатной температуре) и умеренно растворимых соединений, а также адсорбцией жидких ограниченно растворимых или твердых неограниченно растворимых соединений различия не делаются, тогда как на соответствующих изотермах наблюдаются характерные особенности. В первом случав уже при низких концентрациях адсорбция достигает некоторой предельной величины, после чего практически не изменяется (L2) Изотермы адсорбции жидких растворенных веществ часто имеют S-образную форму - адсорбция быстро увеличивается по мере достижения предела растворимости (S3).
5. Теория адсорбции из бинарных растворов
Адсорбцию из растворов следует рассматривать как процесс концентрирования на поверхности одного из двух адсорбирующихся компонентов.
Исходя из фундаментального уравнения Гиббса
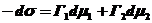 (25)
(25)
где Г1 и Г2 - величина Гиббсовой адсорбции компонентов,
μ1 и μ2 - химический потенциал компонентов,
чтобы связать Гиббсову адсорбцию с концентрацией компонента, необходимо воспользоваться уравнением Гиббса-Дюгема
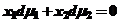 (26)
(26)
где х1 и х2 - мольная доля компонентов.
Тогда:
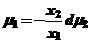 (27)
(27)
подставим в уравнение Гиббса (25) и получим:
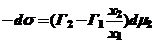 (28)
(28)
Сделав допущение, что общее число молей до и после адсорбции остается неизменным, получим, что при адсорбции одного компонента равномерно увеличивается количество другого. Тогда Г1 =Г2, х2 + х1=1. Из этого получается:
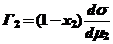 (29)
(29)
и заменив μ2 на с2 получим уравнение, подобное адсорбционному уравнению Гиббса для разбавленных растворов
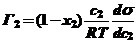 (30)
(30)
Типичным примером зависимости Гиббсовой адсорбции от состава бинарных смесей представлена на рис. 13. Точка пересечения отвечает такому состоянию системы компонентов в целом, при котором одинаковы составы раствора и поверхностного слоя. Это возможно только при следующем допущении: во-первых, первый компонент поверхностно-инактивен для второго и наоборот и, во-вторых, приравняв в поверхностном слое одного компонента равняется убыли второго компонента в этом слое.

 Г2
Г2


 Г2 Г1
Г2 Г1

 Х=1
Х=1
Г1
Рис. 13. Зависимость Гиббсовой адсорбции от состава бинарных растворов
При адсорбции ПАВ из раствора на поверхность твердого тела соблюдается правило Траубе и его обращение в зависимости от природы твердой поверхности и растворителя (рис. 14).
Газ Воздух Газ
|

бензол вода
бензол вода
(неполярный) (полярная)
Граница раздела фаз «газ – жидкость»
|

Уголь Двуокись кремния
(неполярный) (полярная)
Граница раздела фаз «жидкость - твердое тело»
Рис. 14. Схема ориентации ПАВ на границе раздела фаз "газ - жидкость" и "жидкость - твердое тело"
При адсорбции из бинарных растворов наблюдается перераспределение компонентов х1 и х2 между объемом и поверхностным слоем. В общем виде подобный обмен можно представить в виде квазихимической обменной реакции с константой обмена
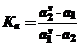 (31)
(31)
где аis и аi - активности компонентов в поверхностном слое и в объеме.
Выразив активность а через мольные доли и коэффициент активности Кf получим:
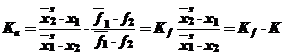 (32)
(32)
где К коэффициент, учитывающий изменение коэффициента активности. Заменив мольные доли первого компонента на мольные доли второго, исходя из выражения (х1 + х2 = 1), получим:
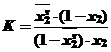 (33)
(33)
Относительно мольной доли второго компонента выражение принимает следующий вид:
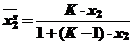 (34)
(34)
Это уравнение называют общим уравнением изотермы адсорбции из бинарных растворов с константой обмена. Константа К - коэффициент разделения компонентов (концентрационная константа) равняется термодинамической константе Ка (31) только в том случае, когда Кf = 1.






![]()
1
3
2
![]()
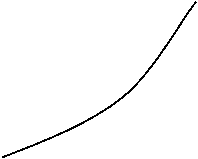
Рис. 15. Изотермы адсорбции из раствора по уравнению (34)
Проведем анализ уравнения (34) (рис. 15).
Если х2 = 0, то при любом значении К можно пренебречь величиной (К-1)х2, и изотерма адсорбции принимает вид закона Генри :
![]() (35)
(35)
Таким образом отклонение от линейной зависимости ![]() от х2 наблюдается при К > 1 (кривая 1), и при К < 1 (кривая 2). С ростом концентрации и значительном отличии К от 1 ( К>>I и К<< I ) можно не учитывать влияние изменения Кf.
от х2 наблюдается при К > 1 (кривая 1), и при К < 1 (кривая 2). С ростом концентрации и значительном отличии К от 1 ( К>>I и К<< I ) можно не учитывать влияние изменения Кf.
Если К>>1, то выражение К-1 имеет положительный знак и уравнение принимает вид уравнения Ленгмюра:
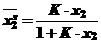 , К''=К (36)
, К''=К (36)
Если К<<1, то выражение К-1 приобретает отрицательный знак и уравнение (34) принимает следующее соотношение:
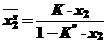 (37)
(37)
Наиболее интересной представляется зависимость изменения знака величины (К-1) по мере изменения состава раствора при адсорбции бинарных систем. Может наблюдаться такой случай, когда при малых концентрациях изотерма проходит выше диагонали (кривая 3 рис. 15) начала координат, а при повышенных концентрациях - ниже диагонали и имеет точку пересечения при К=1. Это соответствует проявлению адсорбционной азеотропии.
Природа твердого тела, растворителя и растворенного вещества во многом определяют избирательность (селективность) адсорбции, которая характеризуется коэффициентом разделения - отношением коэффициентов распределения компонентов на поверхности и в объёме
![]() (38)
(38)
где сs - концентрация компонентов на поверхности,
сv - концентрация компонентов в объеме.
Тогда коэффициент разделения β для бинарных систем имеет следующее выражение:
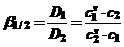 (39)
(39)
и показывает, во сколько раз различаются коэффициенты распределения разделяемых компонентов.
Коэффициенты распределения и разделения не зависят от соотношения между количеством адсорбента и раствора и поэтому не могут характеризовать распределение массы вещества между этими фазами. В этом случае распределение массы вещества целесообразно характеризовать степенью извлечения и степенью разделения. Чем меньше степень извлечения вещества, тем лучше их разделение в поверхностном слое и тем хуже их разделение в объеме раствора.
Для оценки адсорбируемости веществ широко используется правило полярности (1927г.). Это правило заключается в том, что вещество может сорбироваться на поверхности раздела фаз, если в результата его адсорбции будет уравниваться разность полярностей этих фаз. Таким образом по полярности адсорбируемое вещество должно занимать промежуточное положение между твердой поверхностью и растворителем. Наиболее характерными свойствами, определяющими в большинства случаев полярность компонентов является величина диэлектрической проницаемости ξ. (В настоящее время существуют более чувствительные характеристики, особенно для органических веществ. Поэтому адсорбция поверхностно-активного вещества, имеющего диэлектрическую проницаемость ξПАВ возможна в следующих случаях:
ξтв > ξПАВ > ξр-ля или ξтв < ξПАВ < ξр-ля
Из правила уравнивания полярности следует, что чем больше разность полярностей между растворимым веществом и раствором, то есть, чем меньше растворимость вещества, тем лучше оно будет адсорбироваться.
Поэтому дифильные молекулы ПАВ в водном растворе (рис. 14) адсорбируются таким образом (частокол Ленгмюра), чтобы выравнить полярность твердого тела и растворителя.
5. Адсорбция низкомолекулярных несмешивающихся
и смешивающихся жидких систем
В основном при изучении адсорбции из растворов на границе раздела "твердое тело - жидкость" экспериментально измеряется изменение концентрации раствора. Так как раствор содержит более одного компонента изотерма изменения концентрации, по существу, является изотермой изменения состава раствора и для двухкомпонентной системы описывается следующим соотношением:
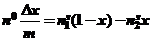 (40)
(40)
где Δх- уменьшение мольной доли компонента,
n0 - мольная доля исходного раствора, приходящаяся на m граммов адсорбента,
n1s и n2s - число молей компонента 1 и 2 в расчете на 1 грамм адсорбента.
Обозначения, символы и терминология приняты ИЮПАК.
Так как в уравнение (40) входят две неизвестные величины, следовательно, индивидуальные изотермы могут быть получены только с использованием дополнительной информации. В этом случае возможны два варианта:
Вариант 1.
Если растворы сильно разбавлены и если растворимость растворенного вещества мала даже при большом n2s произведение n2sx < и n1s(1-x) = n2s (пример: стеариновая кислота в бензоле). Изотерма изменения состава раствора преобразуется в индивидуальную изотерму адсорбции растворенного вещества.
Вариант 2.
Если же изотерма адсорбции имеет максимум при мольной доли растворенного вещества более низкой, чем предел растворимости, что также возможно. В этом случае, несмотря на низкие значения x растворенного вещества, изотерма изменения состава раствора явля ется изотермой адсорбции только растворенного вещества. Можно предположить, что монослой заполняется при относительно низкой концентрации растворенного вещества и ![]() = 0. Уравнение изотермы адсорбции приобретает следующий вид:
= 0. Уравнение изотермы адсорбции приобретает следующий вид:
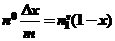 (41)
(41)
Следовательно, изотерма изменения состава раствора после завершения образования монослоя адсорбатом линейно падает с изменением концентрации.
Определение количества растворенного вещества, адсорбированного при определенных условиях - задача несложная, но установить механизм адсорбции и влияние на него возможных переменных факторов намного сложнее и не всегда учтено.
Известно, что на величину адсорбции на твердом теле из растворов первостепенное влияние оказывает поверхность, природа растворенного вещества, его химическое строение и взаимодействие с растворителем, природа самого растворителя, природа взаимодействия между поверхностью и адсорбированным из раствора веществом, структура адсорбированного слоя, его толщина и плотность упаковки, и, конечно, температура.
Четкое представление о природе поверхности исследуемого образца, получение одинаковых поверхностей в различных условиях, характеристика основных свойств твердой поверхности - наиболее трудная задача. Загрязнение под действием окружающей среды оказывается достаточным, чтобы вызвать необратимые изменения в свойствах поверхности. Методы подготовки образцов, хотя и весьма разнообразные, но и они не дают возможность получить поверхности хотя бы с близкими свойствами. Наиболее доступным адсорбентом с наиболее воспроизводимыми свойствами поверхности являются графитовые сажи, а их использование дает надежную информацию при исследовании адсорбции из раствора.
Химические свойства других модификаций углерода значительно отличаются, что связано с наличием на поверхности ряда кислотных и основных групп. Диоксид кремния в результате температурной обработки образует огромное количество пористых и непористых модификаций, отличающихся типом и числом гидроксильных групп. А диоксид титана образует известные модификации - рутил, анатаз и др., имеющие те или иные анионные группы, сказывающиеся на свойствах поверхности.
Так например, при исследовании адсорбции двухосновных кислот (от щавелевой до себациновой) из водного раствора на графитовой саже - сферон 6 (по мере увеличения температуры прокаливания в вакууме и удаления кислотных соединений, определяющих сродство к полярному растворителю) было обнаружено увеличение адсорбции в ряду
сферон 6 (2700°С) > графон > сферон 6 (1000°С) > сферон 6 (без обработки).
Фенол и нитробензол хуже адсорбируются на графоне по сравнению с полициклическими углеводородами в циклогексане, бензол и нафталин сильнее адсорбируются на сфероне 6, чем на менее полярном графоне.
Данные по адсорбции полярных адсорбатов из растворов в углеводородах на поверхности диоксидов убедительно показывают насколько важно знать предысторию образца. Так, на непористых силикогелях с различной степенью гидроксилирования (2,8; 4,2; 7,9 ммоль ОН/м2) показано, что адсорбция алифатических спиртов из разбавленных растворов в тетрахлоруглероде (ССl4) повышается по мере увеличения степени гидроксилирования. Неоднократно отмечалось, что адсорбированная вода на поверхности диоксида кремния и титана в значительной мере способствует уменьшению адсорбции кислот и спиртов из разбавленных растворов в углеводороде.
При анализе данных по адсорбции из растворов пористыми твердыми телами необходимо учитывать как химические, так и физические характеристики доступности поверхности. В ряде случаев, преимущественной адсорбцией обладают молекулы с малым размером, при этом степень и внутренней поверхности определяется отношением размеров пор и размеров молекул. Близость поверхности стенок пор, а также селективность адсорбции, определяемая химией поверхности твердого тела, в основном является следствием более сильной адсорбции внутри пор по сравнению с адсорбцией на внешней поверхности. Разделить оба фактора не всегда предоставляется возможным, поэтому в большинстве случаев интерпретация полученных данных адсорбции из разбавленных растворов пористых твердых поверхностях является до некоторой степени неопределенной.
Особенности индивидуального строения молекул растворенного вещества такие как, наличие полярных трупп, длина цепи, структура кольца, физическое состояние в растворе, дипольный момент, постоянная Гаммета, растворимость вещества в растворителе оказывают существенное влияние на адсорбцию их на поверхности твердого тела.
Соблюдение известного правила Траубе наблюдается только в случае простейших жирных кислот на угле из водных растворов в разбавленных концентрациях. Попытка установить закономерность адсорбции от длины углеводородного радикала спиртов, количества ароматических колец в молекуле, дипольного момента на широком спектре адсорбтивов не увенчалась успехом. Величина адсорбции молекул растворенного вещества определяется доступностью поверхностных групп, образованием "упорядоченного" слоя сольватированных углеводородных цепей и, в основном, не зависит от их длины.
Растворитель в значительной мере определяет адсорбцию растворенного вещества на твердом теле так как, во-первых, их совместная растворимость и отклонение от идеальности зависит от взаимодействия растворенного вещества в растворе, во-вторых, взаимодействием с адсорбатом, зависящим от их химической природы и структуры и, наконец, взаимодействие с растворенным веществом в адсорбционном слое вблизи границы раздела "твердое тело - раствор".
Так как при исследовании адсорбции растворов в большинстве случаев не учитывается адсорбция растворителя на поверхности твердого тела, особенно когда равновесная концентрация раствора велика. Изотерма изменения состава раствора, которая может принимать как положительные, так и отрицательные зависимости, существенно отражает конкурирующую адсорбцию обоих компонентов.
Существует пять классов изотерм (рис. 15).
|

Рис. 16. Классификация изотермы изменения состава по Шаю и Надю.
Ниже приведена типичная изотерма адсорбции, рассчитанная по экспериментальным данным при условии, что адсорбция монослойна, площадь, приходящаяся на моль компонента в адсорбционном слое, не зависит от концентрации адсорбата в растворе, и компоненты раствора не содержатся в твердой фазе (рис. 17).
|

Рис. 17. Изотерма адсорбции в системе этанол (1) и ксилол (2) (рутил при
298 К) и изотерма изменения состава раствора (3)
В настоящее время благодаря хорошей экспериментальной базе, которая позволяет количественно оценить параметры жидкости на молекулярном уровне, стало возможным рассчитать и построить модель границы раздела "твердое тело - раствор", базирующихся либо на решетчатой модели поверхности и раствора, либо на модели однородной "поверхностной фазы", находящейся в равновесии с бесструктурной жидкостью.
Для расчета параметров решеточной модели используют метод Монте-Карло, который в значительной мере углубил наши представления о жидком состоянии. Применяемый в методе Монте-Карло подход требует, чтобы полная внутренняя энергия любого расположения молекул (конфигурации) в системе может быть вычислена или уже известна. Затем конфигурация молекул меняется (здесь применимы строгие правила учета состояния) и рассчитывается разность энергий между новой и старой конфигурациями, и полученная разница анергии позволяет судить о приемлемости этой новой конфигурации и так далее. Таким образом, совокупность принятых конфигураций образует некоторую цепь (цепь Маркова), которая позволяет рассчитать механические свойства каждой принятой конфигурации. Цепь обрывается в тот момент, когда среднее значение рассматриваемых механических свойств не зависит от некоторой выбранной произвольной конфигурации.
Чтобы осуществить расчет по методу Монте-Карло, необходимо знать потенциальную энергию данной конфигурации системы, Если система стационарна, то ее внутреннюю энергию можно разложить, во-первых, на потенциальную энергию взаимодействия с гравитационным и другими внешними полями, во-вторых, кинетическую энергию теплового движения, молекул и, в-третьих, потенциальную энергию взаимодействия между самими молекулами. Средняя, энергия первых двух вкладов получается непосредственно, а внутренняя энергия третьего типа слагается из энергии отталкивания при малых межмолекулярных расстояниях, всегда существующей энергии дисперсионного взаимодействия и электростатического вклада постоянных электрических моментов (заряда, диполей), Выражения потенциальных функций парных межмолекулярных взаимодействий для атомов инертных газов достаточно хорошо известны, а простейший потенциал Леннарда-Джонса удовлетворительно выражает объединенное взаимодействие и определяется следующим образом, представленным при изучении природы адсорбционных сил (21).
Кроме макроскопических свойств системы (давление, плотность, сжимаемость и энергия) методом Монте-Карло рассчитывают и микроскопические свойства - функцию распределения частиц по размеру. Однако, полученные модельные расчеты по методу Монте-Карло значительно отклоняются от реальной структуры жидкости, но разработанная и действующая "решеточная модель жидкости" в настоящее время позволила рассчитать модели жидкой фазы с учетом межмолекулярного взаимодействия, что и было сделано Гуггенгеймом.
Другая модель, основанная на том, что граница раздала фаз может быть аппроксимирована тонким однородным слоем жидкости. Выделенный тонкий слой с одной стороны ограничен твердой поверхностью, а с другой стороны - объемом гомогенного раствора. В этом случае для расчета полной энергии межмолекулярного взаимодействия необходимо знать только соответствующие одночастичные или парные функции распределения, которые существуют в так называемом "липком жестком слое". Рассчитанный потенциал не обладает физическим смыслом, но учитывает действие сил притяжения и отталкивания и позволяет получить реальные макроскопические величины, например, второй вариальный коэффициент.
Сравнение последнего с методом Монте-Карло единственная возможность проверить полученные результаты еще раз, чтобы сказать о перспективном начале термодинамической оценки состояния адсорбционного слоя.
7. Адсорбция поверхностно-активных веществ на твердых поверхностях
Поверхностно-активные вещества (ПАВ) ведут себя в водных растворах иначе, чем большинство других, растворенных в воде веществ. Необходимо напомнить, что ПАВ при очень низких концентрациях находятся в свободном молекулярном состоянии (истинный раствор), а начиная с некоторой критической концентрации (ККМ) происходит агрегация, случай когда индивидуальные молекулы и мицеллярные кластеры существуют в динамическом равновесии. Дальнейшее увеличение концентрации ПАВ в водном растворе мицеллы различного размера и формы концентрируются в характерные симметричные структуры и образуют лиотропные изоморфные фазы. В результате адсорбции ПАВ существенно изменяют свойства границы раздела фаз при весьма низких концентрациях и в следствие кооперативной самоассоциации они агрегируются с образованием мицелл.
Существенно отличается теория адсорбции неионогенных и ионогенных ПАВ на твердом теле.
7.1. Адсорбция неионогенных ПАВ
Рассмотрим наиболее простую адсорбцию неионогенных ПАВ, которую относят к представителям классической физической адсорбции. Неионогенные ПАВ от другого широкого круга ПАВ отличаются тем, что даже малые изменения концентрации, температуры, молекулярной структуры, природа поверхности твердого тела оказывают существенное влияние на их адсорбцию. Это, прежде всего, связано с тем, что взаимодействие “ПАВ – ПАВ” и “ПАВ – растворитель” вызывает агрегацию растворенного вещества в объеме или приводит к изменению ориентации и упаковки ПАВ на поверхности.
На рис. 18 показана схема вероятных изменений ориентации молекул неионогенных ПАВ при адсорбции из водного раствора на твердом теле по мере увеличения концентрации.
На первой стадии адсорбции (а), когда число активных центров и количество адсорбированных молекул ПАЗ очень мало, взаимодействием между молекулами ПАВ можно пренебречь. Адсорбция в основном определяется вандерваальсовыми силами, в большинстве случаев дисперсионными. Взаимодействие между твердым телом и гидрофобной группировкой ПАВ приводит к расположение молекул параллельно поверхности, так как последние адсорбируются положительно. Если молекулы при адсорбции располагаются параллельно поверхности, то энергия адсорбции увеличивается равными порциями с каждым дополнительным атомом в углеводородной цепи. При этом правило Траубе соблюдается для большинства неионогенных ПАВ и твердых поверхностей.
По мере насыщения к монослойному насыщению горизонтально расположенными молекулами (б) наблюдается постепенное уменьшение наклона изотермы адсорбции. Можно предположить, что ПАВ в этом случае вытеснит большую часть молекул растворителя с поверхности, но все таки некоторое его количество остается на активных центрах твердого тела.
Все последующие стадии адсорбции определяются главенствующей ролью взаимодействия молекул ПАВ друг с другом за счет гидрофобного взаимодействия, которая зависит от природы твердой поверхности и гипофильно-липофильного баланса ПАВ (соотношение полярной и неполярной составляющих молекулы).
|

|

Рис. 18. Модель адсорбции неионогенных ПАВ по мере увеличения концентрации ПАВ (а, б, в, г, д) и изотермы адсорбции (в)
Например, в случае 1 мы имели неполярный адсорбент и ПАВ с короткой цепью (низкое ГЛБ). В случае 3, наоборот, преобладание длинноцепочечной молекулы ПАВ с высоким ГЛБ на сильнополярной поверхности. Вариант 2 - некоторое промежуточное состояние.
Третья стадия адсорбции (в), когда концентрация ПАВ в растворе приближается к ККМ, способствует агрегации цепей адсорбированных молекул. Это вызывает вертикальную ориентацию. При этом наблюдается резкое увеличение количества адсорбированных молекул, некоторая десольватация, вытягивание скрученных молекул за счет сжатия головной группы и, в результате этого, значительная плотноупакованность монослоя. На неполярных адсорбентах наблюдали образование полумицелл на поверхности при концентрациях более низких, чем ККМ в объеме раствора. Это объясняется тем, что энергия адсорбции метиленовой группы почти равняется ее энергии мицеллообразования. Однако, на полярных адсорбентах, где полярная головная группа может довольно сильно связывается с поверхностью, а углеводородная цепь отделена от поверхности твердого тела на значительное расстояние, для достижения плотной упаковки понадобится значительно большая концентрация ПАВ в растворе, чем необходимо для ККМ.
Дальнейшее повышение концентрации неионогенного ПАВ в растворе (г, д) приводит к тому, что взаимодействия в адсорбционном слое подобны взаимодействиям в объеме раствора. При этом изменение энтальпии обусловлено установлением равновесия между усиливающимися углеродно-углеродными взаимодействиями или головными группами и процессом разрушения сольватных оболочек. Отмечено, что теплота адсорбции становится постоянной, хотя с увеличением температуры адсорбция растет, так как головные группы становятся более компактными из-за меньшей сольватизируемости.
Проведенные немногочисленные исследования показали, что поверхность твердого тела покрыта плотноупакованными полумицеллами, т. е. агрегатами такого размера и формы, которые можно получить, разделив находящуюся в объеме мицеллу пополам. В ряде случаев отмечалось, что в системах «неионогенное ПАВ - полимерный адсорбент» при концентрациях ниже ККМ адсорбция отсутствует, а молекулы ПАВ удерживаются вблизи твердой поверхности дальнодействующими ван-дер-ваальсовыми силами через толстую прослойку сольватизированного слоя растворителя. Адсорбция в этом случав не происходит до тех пор, пока преобладает влияние поверхностной агрегации. Здесь требуется уточнение. Исходя из того, что большинство неионогенных ПАВ содержит полярную группу, которая достаточно сильно адсорбируется полярными поверхностями, например, за счет водородных связей, так что отсутствие адсорбции при концентрации ниже ККМ является кажущимся и может быть объяснено нечувствительностью эксперимента при низких концентрациях ПАВ.
7.2. Адсорбция ионогенных ПАВ
Адсорбция ионогенных ПАВ на границе раздела «твердое тело – раствор» имеет огромное значение, так как определяет устойчивость коллоидных, систем и регулирует смачиваемость. Способность ПАВ в создании устойчивых коллоидных систем лежит в основе разработки детергентов и антикоагулянтов, фармацевтических и красящих дисперсий, эмульсионной полимеризации и флотации. Смачивание играет важную роль при очистке, диспергировании и флотации, крашении и печатании, при использовании пестицидов и гербицидов.
В процессе адсорбции в случае ионогенных ПАВ участвуют как минимум 4-х компонентная система, а в случае высоких концентраций ПАВ количество отдельных систем может достигать 8-ми, обладающих индивидуальными свойствами. Изменение смачиваемости жидкостью твердой поверхности при добавлении ПАВ определяется не только адсорбцией молекул самого ПАВ, но и адсорбцией двух других, смачивающихся в системе, границ раздела «газ - твердое тело» и «газ – жидкость». Из-за сложностей взаимодействий, которые происходят в подобных многокомпонентных системах, несмотря на большое количество исследований по этому вопросу, единой теории, объясняющей хотя бы удовлетворительно процесс адсорбции ионогенных ПАВ на твердом теле, к настоящему времени окончательно еще не выработано.
Основным фактором, определяющим величину адсорбции ионов ПАВ на любой границе раздела фаз является электрическое взаимодействие в области двойного слоя. Таким образом, объяснить процесс адсорбции можно только в случае, когда известно, чем обусловлено возникновение поверхностного заряда, величины электрокинетического заряда и, конечно, структура двойного электрического слоя (ДЭС).
Существует несколько механизмов образования поверхностного заряда, хотя общий поверхностный заряд из-за наличия в растворе противоионов и электронейтральности системы в целом равен нулю. Наиболее простейшими считаются модели AgI, BaSO4, где вследствие преимущественного растворения одного типа ионов решетки на поверхности кристалла адсорбируются потенциалопределяющие ионы, не влияющие на химический потенциал фазы.
Грэм предложил модель двойного электрического слоя для границы раздела твердое тело - жидкость, которая приведена на рис. 19.
|

Рис. 19. Модель ДЭС по Грэму
Общая плотность заряда σв внешней плоскости Гельмгольца и задней составляет σd, а дифференциальная емкость этой диффузной области равна Cd и параметры двойного слоя связаны между собой следующим комплексом уравнений:
К1 = σ0 . (φ0 – φβ), (42)
К2 = - σd. (φβ – φA), (43)
К-1 = К1-1 + К2-1, (44)
Cd = - dσd/dφA, (45)
σ0 + σв + σd =
В случае симметричного электролита
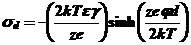 (47)
(47)
где ε - абсолютная диэлектрическая проницаемость объема растворителя,
γ - обратная величина ионной силы раствора Дебая-Гюккеля, которая составляет при Cd
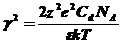 (48)
(48)
z - валентность иона,
е - заряд иона.
Исходя из электрохимического равновесия между числом ионов на поверхности твердого тела и в объеме раствора, выведенного Нернстом, имеем
Δφ = NA(pX), (49)
где Δφ - изменение внутреннего потенциала твердой фазы с изменением концентрации потенциалопределяющего иона типа X с зарядом z,
N = - 2,3 (kT/ze), (50)
Заменив Δφ на φ0, который характеризует изменение поверхностного потенциала вблизи поверхности твердого тела, связывающей ориентированные диполи воды (жидкости) внутри твердой фазы
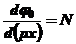 (51)
(51)
В изоэлектрической точке φd = 0 получим уравнение
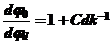 (52)
(52)
Вводя выражение
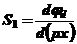 (53)
(53)
и подставляя его в уравнение (52), получим:
NS1-1 = 1 + C d0 k
Так как φd нельзя измерить непосредственно, то для проверки уравнения используют легко экспериментально доступный ζ - электрокинетический потенциал, который с некоторыми допущениями позволяет рассчитать адсорбцию ионов на твердом теле, тогда уравнение приобретает следующий вид
NS-1 = (1 + Cd0k-1) exp (æΔ
В изоэлектрической точке φd = ζ = 0 оценивать Δ1 нет никакой необходимости.
При построении зависимости NS-1 от Cd0 для частиц AgJ в растворах KNO3 имеет вид прямой, отсекающей от оси ординат отрезок единичной длины, соответствующий Δ1, и по расчетам не превышает 0,5 Нм. Это позволяет считать справедливым применение равенства φΔ и ζ при объяснении межфазных явлений, в том числе включая адсорбцию ионогенных ПАВ.
Также предполагают, что механизм возникновения заряда на поверхности неорганических оксидов SiO2, MgO, TiO2, Al2O3 и других определяется присутствием способных к ионизации групп, какими могут быть амфотерные гидроокисные группы.
Механизм возникновения заряда, хотя и значительно идеализированный, можно себе представить следующим образом:
H+ H+
MeОН2+ ↔ MeОН ↔ MeO¯.
В этом случае подобные межфазные поверхности заряжаются потенциалопределяющими ионами Н+ или 0Н¯, что справедливо для ионных решеток, которые подчиняются уравнению Нернста (51). В этом случае линейная зависимость NS-1 от Cd0 для TiO2 и SiO2, позволяет рассчитать, что Δ1 меньше 0,5 нм. Это еще раз подтверждает правильность предложенной модели и для поверхностей оксидов, а также позволяет количественно оценить поверхностные группы, способные к ионизации.
Монофункциональные группы, такие как R–C–SO3¯, R–C–NH2¯ и другие, в результате включения их в процесс полимеризации латексов и присутствии на поверхности вызывают ионизацию, что в свою очередь приводит к возникновению поверхностного заряда. Поверхностный заряд газовой сажи связан с присутствием различных комплексов кислорода, химически связанных с атомом углерода базисных плоскостей и действующих как слабые кислоты. Другим механизмом объясняют образование заряда на поверхности глин и слюды. Эти слоистые силикаты построены из двумерных сеток кремнекислородных тетраидов и вследствие замещения некоторых атомов Si+4 на Al+3, или Al+3 на Mg+2 кристалл приобретает некоторый отрицательный заряд. Особенно это проявляется на ребрах чешуи.
Исходя из построенной модели, число молей адсорбированного ПАВ, приходящихся на единицу поверхности (Г1) зависит от объемной концентрации раствора С1. Форма изотермы не всегда является правильным выводом о характере адсорбции. Для расчета применяют метод конгруэнтности (подобия) адсорбции по отношению к заряду или потенциалу. При этом адсорбция конгруэнтна к заряду, если электрическая составляющая свободной энергии (σ0),выраженная через заряд, является постоянной и в графической зависимости Г1 от С1 наблюдаются параллельные друг другу графики. Составляющие свободной энергии адсорбции в основном зависят от степени заполнения поверхности θ. Наиболее широко применимо описание изотермы адсорбции по уравнению Штерна-Ленгмюра
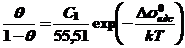 (56)
(56)
где Δσ0адс - стандартная свободная энергия, составляющая энергию адсорбции, не зависящая от степени заполнения.
При низких степенях заполнения (θ → 0) Грем вывел аналогичное уравнение
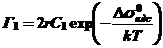 (57)
(57)
где r - радиус адсорбированного иона.
Объяснение различных механизмов адсорбции разнообразных ионогенных ПАВ и адсорбентов включает в себя то, что свободная энергия адсорбции состоит из нескольких составляющих
Δσадс = Δσэл + Δσспец (58)
Δσэл - электрическое взаимодействие, которое в свою очередь складывается из энергии раствора Δσр-ра и дипольного члена Δσдип:
Δσэл = Δσр-ра + Δσдип (59)
или
Δσэл = z1eφl + ∑Δni μi ES, (60)
где Δni - изменение числа адсорбированных диполей i с дипольным моментом μi,
ES - напряженность электрического поля в плоскости адсорбированных частиц.
Принято считать, что Δσдип обусловлен обменными процессами между ионами ПАВ в объеме раствора и в адсорбированном состоянии, происходящих в результате десорбции молекул воды с поверхности. Если пренебречь Δσдип, то можно рассчитать Δσэл во всех возможных вариантах.
I ВАРИАНТ. Если заряд иона ПАВ противоположен по знаку общему объемному заряду (σ0 + σβ), то z1 и φβ имеют противоположные знаки, тогда Δσр-ра < 0 и электрическое взаимодействие способствует процессу адсорбции, соответственно, при низких степенях заполнения отрицательное для катионоактивных ПАВ и положительное для анионоактивных ПАВ.
II ВАРИАНТ. Если заряд иона ПАВ имеет тот же заряд, то z1 и φβ имеют тот же знак, а Δσр-ра > 0. В этом случае электрическое взаимодействие препятствует процессу адсорбции. Однако при высоких степенях заполнения в силу преобладания специфической адсорбции происходит перезарядка в изоэлектрической точке. При этом анионоактивные ПАВ придают поверхности отрицательный заряд, а катионоактивные - положительный.
Ш ВАРИАНТ. Δσэл равна нулю, тогда адсорбция определяется наличием специфической адсорбции Δσспец. Проведенные эксперименты показали, что ионогенные ПАВ в этом случае не адсорбируются на некоторых одноименнозаряженных поверхностях. На рис. 20 показано изменение адсорбции додецилсульфоната натрия на Al2O3 от рН раствора. Адсорбция уменьшается по мере понижения величины положительного поверхностного заряда. Следует заметить, что выше изоэлектрической точки кривые ζ - потенциала совпадают, что говорит об отсутствии процесса адсорбции.
|

Рис. 20. Зависимость ζ - потенциала Al2O3 от рН раствора додецилсульфоната натрия при ионной силе раствора 2.10-3 моль/дм3.
Концентрация ПАВ, моль/дм3: ; , ; 4 - 2,5.10-4
Специфическая адсорбция Δσспец включает в себя все другие вклады в свободную энергию, зависящей от неэлектрической природы системы
Δσспец = Δσцц + Δσцт + Δσпт + …
где Δσцц - изменение свободной энергии при притяжении «цепь – цепь»,
Δσцт - то же, «цепь - твердое тело»,
Δσпт - то же, «полярная группа - твердое тело».
Если «цепь – цепь» можно объяснить гидрофобным связыванием, то «цепь - твердое тело» объясняется Ван–дер–Ваальсовыми дисперсионными взаимодействиями. Характер взаимодействия во многом определяется особенностями структуры ионогенного ПАВ вблизи поверхности твердого тела, поэтому имеет смысл говорить, что Δσцц и Δσцт включают как гидрофобное, так и дисперсионное взаимодействие. При этом Δσцт в значительной мере зависит от природы поверхности твердого тела, ассоциированной структуры адсорбированной воды и от того разрушается или не разрушается эта структура гидрофобными цепями.
Иллюстрацией этого может послужить адсорбция ацетатов алкиламмония, отличающихся длиной углеводородной цепи при рН = 6÷7, приведенная на рис. 21.
|
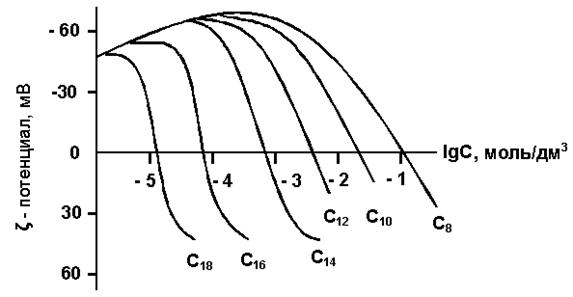
Рис. 21. Изменение ζ - потенциала кварца при рН = 6÷7 от концентрации ацетатов алкиламмония.
Как видно из рисунка адсорбция происходит на поверхности сильно гидратированного кварца и основной вклад в специфическую адсорбцию вносит взаимодействие «цепь – цепь». Эта составляющая приобретает первостепенное значение, так как адсорбирующиеся ионогенные ПАВ стремятся освободиться от водного окружения с образованием на поверхности твердого тела двухмерных агрегатов (полумицелл). До точки, соответствующей ККМ практически не происходит изменение ζ - потенциала поверхности кварца, однако, во всех без исключения случаях адсорбция резко увеличивается вследствие образования полумицелл и резкое изменение ζ - потенциала.
Значение вклада взаимодействия «цепь - твердое тело» Δσцт играет существенную роль на неполярных адсорбентах, которые не вызывают структурирования приповерхностного слоя воды (может быть за исключением самого первого). В то же время, вклад взаимодействия полярная группа - адсорбент наиболее четко проявляется в тех случаях, когда преобладает химическое взаимодействие.
Применение модели двойного электрического слоя для адсорбции ионогенных ПАВ и описание изотермы адсорбции согласно выражениям (56) и (57) возможно только при низких степенях заполнения. Так же как выставленные условия:
1) специфически адсорбируются ионы только ПАВ,
2) один ион ПАВ замещает одну молекулу растворителя, т. е. они имеют одинаковый размер,
3) поверхность считается однородной,
применимы в случае длинноцепочечных молекул ионогенных ПАВ и расстояние как внутри адсорбционного слоя Гельмгольца, так и от слоя Гельмгольца до границы скольжения Δ1 может быть равным только в том случае, когда концентрация ПАВ мала и низка степень заполнения. При этом соблюдается правило Траубе, что показано в табл. 2.
Таблица 2
Величина Δσспец и ККМ анионогенных ПАВ в водном растворе (рН = 2)
на золе нейлона 6-6
ПАВ | - Δσспец / кТ | ККМ, моль/дм3 |
Децилсульфонат С10 | 6,4 | 4, |
Додецилсульфонат С12 | 8,4 | 9, |
Гексадецилсульфонат С16 | 12,9 | 8,С) |
Из таблицы видно, что инкремент в величине Δσспец на одну СН2 - группу в гомологическом ряду увеличивается на 1кТ, что отражает взаимодействие «цепь - твердое тело» при низких степенях заполнения.
Вблизи ККМ изотерма адсорбции ионогенных ПАВ в основном достигает плато, а дальнейшее концентрирование раствора вызывает только рост количества мицелл, которые в этом случае уже не являются поверхностно-активными. Адсорбционный максимум объясняется неоднородностью твердой поверхности, вымыванием из адсорбента легко растворимых веществ, которые солюбилизируются (поглощаются) мицеллами, а также наличием незначительных количеств твердых частичек (центров адсорбции ПАВ) в растворе. Состав мицелл в ряде случаев значительно отличается от состава объема смеси ПАВ, состоящих из молекул различной длины.
Плотноупакованный слой на границе раздела фаз в зависимости от силы взаимодействия «монослой – поверхность» может быть достигнут при концентрациях ионогенного ПАВ как выше, так и ниже ККМ. В случае поверхности углеродного типа плотноупакованный слой молекул ПАВ представляет собой полумицеллу с малой кривизной с вертикальной ориентацией молекул. Однако на поверхности раздела с высокой энергией (сильнополярные) адсорбция углеводородных радикалов вряд ли возможна. Отмечается, что хотя анионогенные ПАВ адсорбируются на твердом теле, но их плотность упаковки адсорбированного слоя определяется электростатическим взаимодействием, ориентацией молекул ПАВ вглубь полислоя, а не полярностью поверхности раздела.
7.3. Пленки Ленгмюра-Блоджетт
Заслуга в установлении мономолекулярной природы слоев на границе раздела «жидкость – газ» принадлежит лауреату Нобелевской премии Ленгмюру, который первый перенес монослои поверхностно-активного вещества с поверхности воды на твердые подложки. В дальнейшем Блоджетт усовершенствовала методику получения моно - и мультимолекулярных слоев ПАВ и разработала технологию переноса их на твердую поверхность. Taк родилось «молекулярное зодчество», которое в настоящее время широко используется при молекулярном (надмолекулярном) конструировании, «молекулярной инженерии», молекулярной и биоподражательной электронике, создании "синтетических" организмов, а также создании молекулярного ансамбля с заранее заданной архитектурой для определенных электрических и оптических характеристик.
В качестве таких молекул широко используются жирные кислоты и их соли (мыла), эфиры, фосфолипиды для получения биослойных мембран, красители, ароматические соединения слоистой структуры и автокомплексы.
Суть метода создания пленок Ленгмюра-Блоджетт на водной поверхности состоит в том, что с помощью плавучего барьера происходит уменьшение площади поверхности, занимаемой ПАВ. При этом дифильные молекулы ПАВ структурируются из газообразных пленок (I стадия) в жидкие (2 стадия) и жидкокристаллические (3 стадия) (рис. 23)
Стадия 1 Стадия 2
![]()


![]()
![]()
![]()
![]()
 2 1
2 1
![]()
![]()
![]()
![]()

![]()
![]()
![]()
![]()

![]()
![]()

![]()
![]()
![]()
![]()

![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]() 3
3


![]()

![]()

![]()
![]()
![]()
![]()
![]()
 Стадия III
Стадия III
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()

![]()
![]()
![]()
![]()
![]()
![]()
![]() 5
5
Рис. 23. Ориентация молекул ПАВ на поверхности воды в зависимости от площади, занятой монослоем (метод Ленгмюра)
1-барьер; 2-блок; 3-груз; 4-механизм подъема и опускания; 5-поверхность.
Последовательность фазовых переходов или полиморфизм монослоев исследуют, наблюдая за соотношением между величиной поверхностного давления (π) и площадью (S), приходящейся на одну молекулу монослоя.
На рис. 24 показан сложный пример полиморфизма азокрасителя, при этом конформационное состояние молекулы меняется по мере увеличения поверхностного давления.
Дальнейшее увеличение поверхностного давления приводит к “сминанию” монослоев или коллапсу и может образоваться двух, трех и четырехслойная структура.
π
![]()

 , мН/м
, мН/м
![]() - азофрагмент
- азофрагмент
![]() - эфирные группы
- эфирные группы
 - углеводородная цепочка
- углеводородная цепочка
Рис. 24. Полиморфизм монослоев ПАВ азокрасителя
Перемещением (мм в минуту) твердой поверхности (5) (рис. 23, стадия 3) через поверхность монослоев опусканием или подниманием обеспечивается различная ориентация ПАВ, изображенная на рис. 25. Обычно различают три типа слоев: два полярных X и Z и один симметричный, неполярный слой Y.

![]()
![]() X Y Z
X Y Z