


Партнерка на США и Канаду по недвижимости, выплаты в крипто
- 30% recurring commission
- Выплаты в USDT
- Вывод каждую неделю
- Комиссия до 5 лет за каждого referral
ГОСУДАРСТВЕННОЕ УЧЕРЕЖДЕНИЕ «ДНЕПРОПЕТРОВСКАЯ МЕДИЦИНСКАЯ АКАДЕМИЯ МИНИСТЕРСТВА ЗДРАВООХРАНЕНИЯ УКРАИНЫ»
“Пересмотрено и утверждено” на методическом заседании кафедры факультетской педиатрии и медицинской генетики Протокол № 1 “29” августа 2014 г. |
Заведующий кафедрой д. м.н., профессор |
МЕТОДИЧЕСКИЕ РЕКОМЕНДАЦИИ К
ПРАКТИЧЕСКОМУ ЗАНЯТИЮ ПО МЕДИЦИНСКОЙ ГЕНЕТИКЕ ДЛЯ СТУДЕНТОВ
V КУРСА
Учебная дисциплина | Медицинская генетика |
Модуль № 1 | Медицинская генетика |
Модуль Содержательный № 4 | Митохондриальные болезни. Болезни с наследственной предрасположенностью.
|
Тема занятия 6 | Общая характеристика митохондриальной патологии. Клиника, диагностика лечения. Болезни с наследственной предрасположенностью. Общая характеристика мультифакториальных заболеваний. Методы профилактики. |
Курс | V |
Факультет | Медицинский |
Актуальность темы. Митохондрий больше всего в энерготропных органах - мозге, сердце, печени, скелетной мускулатуре, почках, эндокринной и респираторной системах, поэтому в первую очередь, страдают именно эти органы и системы вместе или поочередно. Митохондриальные заболевания генетически гетерогенны и клинически полиморфны. Раннее начало заболевания приводит к более тяжелому течению, учитывая то, что начало проявлений митохондриальных заболеваний совпадает с патологическим накоплением мутантной ДНК и протекает прогрессирующе быстро.
В зависимости от того, какой орган поражается, пациенты могут предъявлять жалобы на нарушение моторного контроля, мышечную слабость и мышечные боли, как локализованные, так и разлитые, гастроинтестинальные нарушения (рвота, диарея с признаками экзокринной недостаточности поджелудочной железы) и нарушение глотания, осиплость голоса, связанную со слабостью голосовых связок, задержку роста, сердечные заболевания в большом разбросе от пролапса митрального клапана до различных вариантов кардиомиопатий, формирование диабета, печеночных заболеваний, включающих в себя гепатомегалию или различные варианты аутоимунного идиопатического гепатита, у таких больных возможно появление судорог или эпи-эквивалентов, с последующим формированием эпилепсии; проблем со слухом (нейросенсорная глухота или тугоухость), зрением (чаще всего изменения, связанные со зрительными нервами, в частности, пигментный ретинит); респираторные нарушения (следует помнить, что первичным проявлением дистресс - респираторного синдрома, могущего привести к внезапной смерти ребенка или взрослого, являются повторные эпизоды апноэ у детей, храп и периодический цианоз носогубного треугольника при эмоциональном напряжении), лактат ацидоз, который без лечения может привести к ацидотической коме, общие нарушения развития, а также склонность к частым респираторным заболеваниям (различные виды нарушения иммунитета).
К наиболее типичным проявлениям митохондриопатий относят следующий сиптомокомплекс:
· миопатия, полимиозит;
· офтальмопатия;
· энцефалопатия;
· гепатомегалия;
· кардиомегалия;
· эпилепсия;
· сахарный диабет.
Чаще всего пациенты, страдающие митохондриальными заболеваниями, предъявляют следующие жалобы:
· мышечная слабость, повышенная утомляемость, синдром «вялого ребенка», синдром хронической усталости, непереносимость физических нагрузок;
· головные боли, эпизоды потери сознания и судорожных приступов, утрата ранее приобретенных навыков, деменция;
· ацидотическая рвота, коматозные состояния;
· скелетные нарушения (нанизм);
· нарушение зрения, слепота, офтальмоплегия;
· нарушение слуха;
· кардиалгия, миалгия.
При обследовании таких пациентов выявляют следующие изменения:
· повышенный уровень лактатдегидрогеназы;
· повышенный уровень щелочной фосфатазы;
· повышенный уровень креатинфосфокиназы;
· гипогликемию;
· гематурию;
· повышение СОЭ;
· рабдомиолиз (выявление феномена «рваных» красных волокон RRF при световой микроскопии биоптата мышц);
· лактат-ацидоз.
Классификация митохондриальных заболеваний по типу мутаций в мтДНК (классификации Wallace, 1992 г.):
ТИП МУТАЦИИ | ЗАБОЛЕВАНИЯ |
1. миссенс-мутации
| - нейроофтальмопатия Лебера (LHON); - пигментный ретинит |
2. мутации в генах т-РНК | - синдром MERF (миоклонус-эпилепсия, синдром «рваных красных волокон»; - синдром MELAS (митохондриальная энцефаломиопатия, лактат-ацидоз, инсультоподобные эпизоды) |
3. делеции или дупликации участков мтДНК | - наружная офтальмопатия; - синдром Кернса-Сейра (KSS синдром); - синдром Пирсона (рефрактерная сидеро-бластная анемия с вакуолизацией клеток костного мозга и экзокринной дисфункцией поджелудочной железы); - асимметричный птоз; - двусторонний птоз, сочетающийся с офтальмопарезом и слабостью мышц нижних конечностей; - дилатационная кардиомиопатия; - NARP – синдром (нейропатия, атаксия и пигментный ретинит) |
4. мутации, снижающие число копий мтДНК | - летальная инфантильная дыхательная недостаточность; - синдром молочно-кислого ацидоза. |
5. мутации в ядерной ДНК | - фумаровая ацидемия; - глутаровая ацидемия; - дефицит ацил-КоА-дегидрогеназы жирных кислот с длинной дыхательной цепью; - дефицит 3-гидроксиацил-КоА-дегидро-геназы жирных кислот с длинной дыхательной цепью; - дефицит 3-гидроксиацил-КоА-дегидро-геназы жирных кислот со средней дыхательной цепью; - дефицит 3-гидроксиацил-КоА-дегидро-геназы жирных кислот с короткой дыхательной цепью; - подострая некротизирующая энцефало-миопатия Лея; - прогрессирующая склерозирующая полио-дистрофия Альперса; - трихополидистрофия Менкеса. |
Общая цель – уметь определять основные признаки генных наследственных заболеваний, диагностические критерии отдельных нозологических форм с разными типами наследования.
Конкретные цели обучения:
1. распознавать клинические проявления следующих митохондриальных заболеваний: MELAS, MERRF, MNGIE, Кернса-Сейра, Лея и других;
2. определить необходимость дополнительного обследования больного, включая биохимическое, инструментальное и молекулярно-генетическое, на основе общих признаков митохондриальных заболеваний.
Цели исходного уровня знаний-умений:
1. определение общих вопросов этиологии, патогенеза, генетики митохондриальных заболеваний, их классификации;
2. выявление отдельных нозологических форм митохондриальной патологии на основе сомато-генетического обследования, клинико-генеалогического и синдромологического анализа;
3. интерпретация данных основных лабораторных и специальных методов обследования (биохимических, инструментальных, молекулярно-генетических) митохондриальных заболеваний;
4. определение методов профилактики и лечения (патогенетического и симптоматического) изученных митохондриальных заболеваний.
Чтоб выяснить, отвечает ли исходный уровень Ваших знаний-умений необходимому, выполните такие задания. Правильность решения задач проверьте, сопоставивши с эталоном.
Задания для самоподготовки и самокоррекции исходного уровня умений.
Задание 1.
Какие наиболее частые симптомы характерны для митохондриальных заболеваний?
A. Сколиоз, кифоз, аневризма аорты, вывих хрусталика, арахнодактилия.
B. Непереносимость глюкозы, катаракта, нарушение стула, умственная отсталость.
C. Прогрессирующая миопатия, кардиопатия, нарушения зрения.
Задание 2.
У ребенка есть жалобы на рвоту, ухудшение состояния на фоне инфекционных заболеваний, запах ацетона изо рта во время приступов, нарушение стула, генерализованные судороги на 2-ые сутки жизни в виде, апноэ, прогрессирующая мышечная слабость. В фенотипе задержка роста, черепно-лицевые дизморфии. При обследовании: диффузные изменения паренхимы печени, холестаз, холангит, метаболические изменения в почках; частичная атрофия зрительных нервов, гипоплазия коры головного мозга, некоторая сглаженность извилин; судорожный синдром; в биохимических анализах: повышение уровня общего холестерина, снижение уровня глюкозы, общего белка, повышение АСТ, АЛТ, снижение уровня глутамина, тирозина, повышение уровня треонина в крови, генерализованная гипоаминоацидурия. При молекулярной диагностике обнаружен полиморфизм 8860G в гене тРНК-лизин. Ваш диагноз?
A. частично ядерная митохондриопатия
B. фенилкетонурия
С. Последствия острого инфекционного процесса
Задание 3.
У женщины синдром MELAS, найдена соответствующая мутация. Супруг здоров. Какой прогноз здоровья будущего ребенка в этой семье?
A. Все дети будут здоровыми.
B. 50% здоровых, 50% носителей
C. Все дети унаследуют материнскую мутацию.
Информацию, необходимую для пополнения базисных знаний-умений, можно найти в таких источниках:
Бочков генетика: Учебник – 2-е изд., перераб. И доп. – М.:ГЄОТАР-МЕД, 2001. – С. Мутовин клинической генетики. Учебное пособие. – М.: Высшая школа, 2001. – С. 120-138.После усвоения необходимых знаний выучите следующий материал:
1. «Медицинская генетика» Под ред. чл-корр. АМНУ, проф. , проф. , проф. . Учебник для студентов высших медицинских заведений ІІІ-ІV уровней аккредитации. Киев “Медицина” 2007. 534 с. Рекомендовано МОЗ Украины.
2.Гречанина клинической генетики. – Харьков: «КВАДРАТ». – 2003. – 420 с.
3., , Блинникова синдромы и медико-генетическое консультирование. Атлас-справочник. Изд. 2-е дополн. – М.: Практика, 1996. – 416 с.
Основные теоретические вопросы темы:
Введение. Общая характеристика митохондриальной патологии. Удельный вес митохондриальных болезней в структуре заболеваемости и смертности. Частота и распространенность на разных контингентах. Этиология и патогенез. Различие проявления мутаций митохондриального и ядерного генома на разных уровнях (клинический, биохимический, молекулярный). Пре - и постнатальная реализация действия аномального гена.
Классификация митохондриальной патологии. Мутации в ядерных (частично митохондриальных) генах, мутации в митохондриальных генах, вторичные митохондропатии. Разбор конкретных нозологических форм.
Синдром MELAS. Генетика, характеристика мутаций, клиническая картина, клиническая диагностика, молекулярно-генетические методы диагностики болезни, тактика ведения. Профилактика осложнений.
Синдром MERRF. Генетика, характеристика мутаций, клиническая картина, клиническая диагностика, молекулярно-генетические методы диагностики болезни, тактика ведения. Профилактика осложнений.
Синдром MNGIE. Генетика, характеристика мутаций, клиническая картина, клиническая диагностика, молекулярно-генетические методы диагностики болезни, тактика ведения. Профилактика осложнений.
Синдром Лея. Генетика, характеристика мутаций, клиническая картина, клиническая диагностика, молекулярно-генетические методы диагностики болезни, тактика ведения. Профилактика осложнений.
Синдром Кернса-Сейра. Генетика, характеристика мутаций, клиническая картина, клиническая диагностика, молекулярно-генетические методы диагностики болезни, тактика ведения. Профилактика осложнений.
Демонстрация и разбор больных с митохондриальной патологией.
Принципы диагностики: клиническое исследование, синдромологический анализ, специальные методы – биохимические, ультразвуковые, электрофизиологические, молекулярно-генетические и другие.
Организационная структура занятия:
1. Введение. 5 мин.
2. Этиология, патогенез митохондриальных заболеваний 10 мин.
3. Классификация митохондриальной
патологии 5 мин.
4. Разбор конкретных нозологических форм 45 мин.
5. Демонстрация и разбор больных с митохондриальной
патологией 15 мин.
6. Учебный контроль и коррекция знаний 10 мин.
7. Заключение 5 мин.
Короткие методические указания к работе на практическом занятии.
В начале занятия будет проведен тестовый контроль исходного уровня знаний. Потом – самостоятельная работа студентов с больными. Под руководством преподавателя будет проведен клинический разбор генетических карт больных с митохондриальной патологией. В конце занятия – итоговый тестовый контроль.
Технологическая карта проведения занятия
№ п/п | Этап | Время, минуты | Учебные пособия | Место проведения занятия |
1 | Определение начального уровня | 15 | Задачи | Учебная комната |
2 | Тематический разбор материала, больных с митохондриальной патологией | 60 | Генетические карты, каталоги, фотографии больных, алгоритмы | Учебная комната |
3 | Подведение итогов | 15 | Задачи | Учебная комната |
Граф логичной структуры темы: «Митохондриальные заболевания»
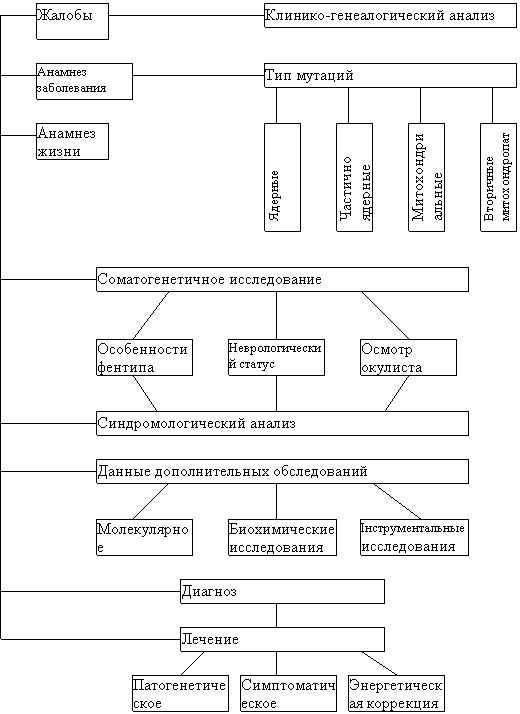 |
Решите несколько заданий-моделей, используя
граф логичной структуры темы
Задача 1.
Пробанд Д, имеет жалобы на боли в грудном отделе позвоночника, быструю утомляемость, неуверенность и шаткость походки, боли в области живота, частые простуды. От первой беременности, первых родов в срок, нормального роста и веса, с двукратным обвитием пуповины. Выявлена дисплазия тазобедренных суставов, подвывих левого сустава, асимметрия бедер, вальгусная деформация нижних конечностей, плоскостопие, левосторонний поясничный сколиоз I-II ст. В результатах обследования: повышение уровня лактата; гипераминоацидемия, с преимущественным повышением аланина, глицина, ФА (трип, тир.); гипераминоацидурия, снижение уровня Р, повышение оксипролина в моче; при УЗИ: периваскулярная инфильтрация в печени, ДЖВП, дополнительная долька селезенки, венозное полнокровие паренхиматозных органов. В почках метаболические изменения; пролабирование створок митрального клапана первой степени, дополнительная хорда в просвете левого желудочка. МРТ головного мозга (атрофический процесс лобно-теменных долей, затруднение ликворной циркуляции), изменения на ЭЭГ (острые волны), ЭМГ (снижение Н-ответа с нижних конечностей) на фоне первичного поражения соединительной ткани. Фенотип: снижение мышечного тонуса, птоз век, признаки соединительно-тканной дисплазии, атаксия. Молекулярная диагностика – полиморфизм мтДНК. Ваш диагноз?
A. НБО соединительной ткани
B. Полинейропатия
С. ГЦУ
D. Митохондропатия
Задача 2
Ребенок с жалобами на резкое ухудшение походки, нарушение речи, двигательная расторможенность, эмоциональная неустойчивость, капризность, мышечная слабость. Задержка темпов психомоторного развития отмечается с 8 месяцев, состояние ухудшилось после перенесенного ОРВИ.
От второй беременности с угрозой прерывания. Роды в срок, нормальный рост и вес. В фенотипе: гиперпигментация в области локтей, колен, гипертрихоз, сухость кожи, аллергическая сыпь, мышечная гипотония, гипомимия, контрактура голеностопных суставов S>D, походка паретическая с выраженной туловищной атаксией. При обследовании: ЯМРТ – негрубо расширены тела боковых желудочков, УЗИ – диффузные, реактивные изменения паренхимы печени; панкреатопатия. При молекулярной диагностики - полиморфизмы митохондриальной ДНК. Ваш диагноз?
A. митохондриальная болезнь.
B. СТД
C. Нейрофиброматоз
Задача 3.
Ребенок 10 л, с жалобами на гипервозбудимость, эмоциональная лабильность, головные боли, энурез, сильная потливость, боли в ногах, быстрая утомляемость, быстрая усталость при ходьбе, частые кровотечения, повышенное артериальное давление. В анамнезе осложнения периода новорожденности - гипоксическое поражение ЦНС, конъюгационная желтуха. В фенотипе асимметрия мимической мускулатуры, склонность к диффузной мышечной гипотонии, переразгибание в коленных суставах, «крыловидные лопатки». При обследовании: повышение уровня лактата, на УЗИ – гепатомегалия, на ЯМРТ – участки демиелинизации. В молекулярно-генетическом исследовании полиморфизм митохондриальной ДНК. Ваш диагноз?
А. Синдром Элерса-Данлоса со вторичной митохондропатией.
В. Синдром MELAS.
C. ФКУ
Дополнение 1
Алгоритм диагностики митохондропатий

![]() Данные родословной Материнский тип наследования
Данные родословной Материнский тип наследования

![]()
![]()
 1. Эпилепсия характер миоклонусы
1. Эпилепсия характер миоклонусы
 2. Миопатия электромиография первичный миопатический
2. Миопатия электромиография первичный миопатический
![]() характер поражения
характер поражения

![]() 3. Кардиомиопатия данные родословной материнский тип
3. Кардиомиопатия данные родословной материнский тип
![]() наследования
наследования
 4.Офтальмопатия данные родословной материнский тип
4.Офтальмопатия данные родословной материнский тип
наследования

![]() МИТОХОНДРИАЛЬНАЯ ПАТОЛОГИЯ
МИТОХОНДРИАЛЬНАЯ ПАТОЛОГИЯ
Дополнение 2
Наиболее частые клинические признаки (органопатии) при митохондропатиях:
ЦНС: поражение мозга, пре-, перинатальная энцефалопатия в виде дегенеративных процессов в мозге - глиоз, гипотрофия, судорожный синдром – миоклонусы, эпи-эквиваленты, резистентная к терапии эпилепсия, полинейропатия, патологические рефлексы, снижение чувствительности, летаргия, кома, задержка психомоторного развития, деменция, атаксия, дистония, "метаболический удар", снижение размеров турецкого седла.
Глаза: птоз, амблиопия, офтальмоплегия, пигментный ретинит, атрофия зрительных нервов, нистагм, катаракта.
Сердце: кардиомиопатия (гипертрофическая), аритмии, нарушение проводниковой системы сердца.
Печень: прогрессирующая печеночная недостаточность (особенно у младенцев), умеренная гепатомегалия, неоднородность паренхимы печени.
Селезенка: спленомегалия, неоднородность паренхимы селезенки.
Почки: тубулопатия (синдром Фанкони), нефрит, почечная недостаточность, пиелэктазия, гидрокаликоз.
Желудочно-кишечный тракт: рецидивирующая рвота, диарея, атрофия ворсин, нарушение экзокринной панкреатической функции.
Эндокринная система: низкий рост, сахарный диабет.
Костный мозг: панцитопения, макроцитарная анемия.
Кожа: раннее старение, недостаточное развитие подкожно-жировой клетчатки.
Скелет: аномалии развития.
Кроме того, прогрессирующий тип заболевания, лактат-ацидоз и специфический фенотип: низкий рост, тонкие волосы, голубые склеры, высокое небо.
Дополнение 3
Синдром MERRF
(миоклонус эпилепсия, “красные рваные волокна”).
Впервые описан N. Fukuhara и соавторами в 1980 году. Синдром MERRF обусловлен точечными мутациями в гене лизиновой тРНК в позициях 8344 и 8356 мтДНК. Заболевание наследуется с внутрисемейным полиморфизмом, что может быть обусловлено разным соотношением между мутантными и нормальными мтДНК в разных ооцитах. Возраст больного при начале заболевания вариабельный от 3 до 65 лет. Ранними клиническими признаками есть быстрое утомляемость при физических нагрузках, боли в икроножных мышцах, снижение памяти, внимания. Наиболее типичным является симптомокомплекс: прогрессирующая миоклонус-эпилепсия, которая включает миоклонус (внезапное, быстрое, краткосрочное мышечное сокращение, которое обусловлено втягиванием ЦНС в патологический процесс), атаксия и деменция. Также у больных отмечаются генерализованные тонико-клонические судороги, нейросенсорная глухота, атрофия зрительных нервов, умеренные признаки миопатии, сенсорные нарушения (разлад вибрационной чувствительности и мышечно-суставного чувства) и другие неврологические симптомы (отсутствие сухожильных рефлексов). Возможно развитие развитие липоматоза. Течение заболевания прогрессирующее.
Критерии диагноза:
· материнский тип наследования;
· дебют в возрасте 3-65 лет;
· поражение ЦНС: миоклонус, атаксия, деменция в сочетании с нейросенсорной глухотой, атрофией зрительных нервов, нарушением глубокой чувствительности;
· лактат – ацидоз;
· умеренное повышение белка в ликворе;
· недостаточность 1, 3, 4 комплексов дыхательной цепи;
· ЭЭГ: генерализованные комплексы “ спайк-волна”;
· ЭМГ: первично-мышечный тип поражения;
· КТ: атрофия мозга, лейкоэнцефалопатия, иногда кальцификация базальных ганглиев;
· “рваные красные волокна” в биоптатах костных мышц; прогрессирующее течение.
Дифференциальный диагноз проводится с другими прогрессирующими миоклонус-эпилепсиями (денторубропаллидольюисовой атрофией, болезнью Гоше, галактосиалидозом ІІ типа, синдромом миоклонуса с почечной недостаточностью и др.), болезнями накопления (например, болезнь Краббе), синдромом Айкарди и другими дизгенезиями мозга. Лечение, вначале, направлено на коррекцию нарушений энергетического обмена, уменьшение лактат-ацидоза, предупреждение повреждений мембран митохондрий свободными радикалами. Установлена эффективность рибофлавина, никотинамида, цитохрома С и коэнзима Q10, L-карнитина, витамина С.
Большое значение имеет также противосудорожная терапия. Препаратами первой очереди являются вальпроаты по 30мг/кг/сутки, а при их неэффективности – клоназепам.
Синдром MELAS
(Митохондриальная энцефалопатия, лактат-ацидоз, инсультоподобные эпизоды)
Синдром впервые был описан S. G. Pavlakis и соавт. в 1984 г. В основе патогенеза синдрома MELAS лежат точечные мутации мтДНК (в позициях 3243, 3271 п. н.), причем есть корреляция между степенью мутации и характером течения заболевания. Для проявления заболевания необходимо накопление определенного количества мутантной мтДНК (56-95%), при этом в одной семье редко встречаются 2 ребенка с классическим вариантом болезни. Первые признаки заболевания появляются, как правило, в 6-10 лет (возможны варианты от 2 до 40 лет). До манифестации заболевания 90-100% больных симптомов заболевания не имеют. Частыми начальными клиническими симптомами являются судороги, рецидивирующая головная боль, рвота, анорексия. Одним из важных симптомов митохондриальной патологии есть непереносимость физических нагрузок (резкое ухудшение самочувствия, появление мышечной слабости, миалгии). Инсультоподобные эпизоды проявляются рецидивирующими приступами головной боли, головокружение, развитием очаговой неврологической симптоматики, коматозными состояниями. Причиной таких “метаболических инсультов” есть острая недостаточность энергетических субстратов в клетках, а также высокая чувствительность сосудов мозга к токсичным воздействиям. Провоцирующими факторами бывает лихорадка, интеркуррентные инфекции.
Судороги – также один из ведущих манифестных симптомов синдрома MELAS, однако они очень вариабельные – фокальные пароксизмы, генерализованные тонико-клонические приступы, миоклонии. Такие эпилептические приступы резистентны к антиконвульсантной терапии. С развитием заболевания развивается деменция. Возможно поражение эндокринной (сахарный диабет, гипопаратиреоз) и сердечно-сосудистой системы (АВ-блокада).
Также отмечаются низкорослость; нарушение зрения, атрофия зрительных нервов; лихорадка; мозжечковый синдром; синдром Вольфа-Паркинсона-Уайта, нарушения сердечной проводимости, прогрессирующая внешняя офтальмоплегия; сахарный диабет.
При синдроме MELAS отмечается поражение комплексов дыхательной цепи. Течение болезни прогрессирующее. При раннем дебюте болезни течение более злокачественное.
Критерии диагноза:
· материнский тип наследования;
· возраст манифестации – до 40 лет;
· мигренеподобная головная боль с тошнотой и рвотой;
· инсультоподобные эпизоды;
· судороги;
· в крови: лактат-ацидоз;
· в моче: повышение уровня органических кислот;
· кальцификация базальных ганглиев на КТ;
· “рваные красные волокна” в биоптатах костных мышц;
· прогрессирующее течение.
Синдром MELAS необходимо дифференцировать от других митохондриальных заболеваний, синдрома Лея (подострая некротизирующчая энцефалопатия), органических ацидемий, гомоцистинурии, синдрома Фабри, врожденных пороков сердца, сосудистых аномалий.
Терапия должна быть направлена на коррекцию биохимического дефекта (коэнзим Q10 по 80-300 мг/день, рибофлавин (100 мг/день), никотинамид (до 1 г/день), дихлорацетат натрия (25-100 мг/кг), витамины К1 (по 25 мг/день), К3 (по 75 мг/день), С (по 2-4 г/день), янтарная кислота до 6 г/день, витамин С (300-500 мг/день), идебенон (90-180 мг/день), цитомак по 4,0 мл в/м. Проведение этой терапии уменьшит проявления симптомов поражения нервной, эндокринной систем и нормализует соматический статус.
Синдром Кернса-Сейра впервые был описан в 1958 г. под названием “пигментный ретинит, прогрессирующая внешняя офтальмоплегия, полная блокада сердца”. Делеция мтДНК была выявлена с помощью молекулярно-генетических исследований только в 1989 г. Как и все митохондриальные заболевания, он имеет неменделирующий характер наследования, высокую частоту спорадических случаев. Это объясняется двумя моментами:
1) большие перестройки мтДНК происходят после оплодотворения яйцеклетки и определяются в основном в соматических, а не в половых клетках;
2) ооциты, которые содержат делеции мтДНК, в большинстве случаев неспособны к развитию эмбриона.
Заболеваемость не зависит от пола, возраст манифестации 4-18 лет.
В клинической картине характерна триада признаков:
· Начало заболевания в возрасте до 20 лет;
· прогрессирующая внешняя офтальмоплегия;
· пигментный ретинит;
· Атриовентрикулярная блокада сердца, мозжечковый синдром,
· повышение уровня белка в ликворе (более 1 г/л).
Птоз, как правило, симметричный и билатеральный; движение глазных яблок резко ограничено. Часто снижается острота зрения, на глазном дне определяется пигментная грануляция. Возможна диплопия, которая не корректируется антихолинестеразными препаратами.
Миопатический синдром определяется через несколько лет после возникновения птоза. Лицо – маскоподобное, гипомимичное, изменяется тембр голоса, частые поперхивания, утомляемость при длительном разговоре.
При физических нагрузках могут развиваться миалгии, крампи, миотония, интенционный тремор. Отмечают эпизоды коми, вследствие метаболических нарушений. Эндокринные нарушения вариабельные (возможна низкорослость, гинекомастия, гипогонадизм, сахарный диабет, гиперальдостеронизм, гипопаратиреоз, дефицит гормона роста). Могут отмечаться кифосколиоз, краниосиностоз, метафизарная дисплазия; нарушения эмалегенеза; поражение почек по типу почечного тубулярного ацидоза или синдрома де Тони-Дебре-Фанкони. Возможны два варианта синдрома – полный и неполный. Полный вариант включает в себя хроническую прогрессирующую внешнюю офтальмоплегию, пигментный ретинит и атриовентрикулярную блокаду. Неполный, в свою очередь, разделяется на два варианта. Первый включает в себя хроническую прогрессирующую внешнюю офтальмоплегию, миопатию нисходящего типа. Второй тип характеризуется только изолированной хронической прогрессирующей внешней офтальмоплегией, встречается редко, имеет поздний дебют. Течение заболевания прогрессирующее.
Критерии диагноза:
· дебют заболевания в возрасте 4-18 лет;
· мозжечковый синдром с интенционным тремором;
· снижение интеллекта;
· прогрессирующая внешняя офтальмоплегия;
· пигментный ретинит, иногда диплопия;
· АВ-блокада сердца;
· белок в ликворе больше 1 г/л ;
· ЭЭГ: неспецифические изменения;
· ЭНМГ: первично мышечный тип нарушения;
· КТ: атрофия коры мозга, лейкоэнцефалопатия, кальцификаты базальных ганглиев;
· в крови: повышение аланина, снижение общего карнитина, фолиевой кислоты, лактата, пирувата;
· “рваные красные волокна” в биоптатах мышечной ткани.
Дифференциальный диагноз: необходимо проводить с другими формами прогрессирующих миопатий (окулофарингодистальной миопатией, супрануклеарным латеральным параличом взгляда со сколиозом и др.), а также с заболеваниями, которые сопровождаются птозом (миастения, диабетическая полиневропатия, синдром Толоза-Ханта, офтальмоплегическая мигрень).
Лечение.
Гипоуглеводная диета, убихинон до 120 мг/день (2мг/кг), длительностью 3-6 месяцев; тиамин до 900 мг/день; L-карнитин по 100 мг/кг/день; фолиевая кислота по 1 мг/день; витамин С по 2-4 г/день; рибофлавин по 50-60 мг/день; витамин Е до 300-500 мг/день. В случае полной АВ-блокады рекомендуется искусственный водитель ритма.
Подострая некротизирующая энцефаломиелопатия Лея
Впервые описана D. Leigh в 1951 году. Подострая некротизирующая энцефаломиелопатия Лея – болезнь первого-второго года жизни (редко манифестирует до 7 лет). Тип наследования – аутосомно-рецессивный, Х-сцепленный рецессивный, митохондриальный.
Критерии диагноза:
· манифестация со второй недели жизни до 7 лет;
· ЦНС: атаксия, задержка психомоторного развития, или регрессия;
· мышечная гипотония или спастичность;
· атрофия зрительных нервов, пигментный ретинит, офтальмоплегия, нистагм;
· респираторный дистресс-синдром;
· пигментный ретинит;
· Рейе-подобный синдром;
· лактат-ацидоз;
· острое истощение после обычных инфекций;
· ЯМРТ: симметричное поражение базальных ганглиев, спонгиозная дегенерация среднего мозга, базальных ганглиев;
· генетический дефект: мутации мтДНК 8993Т>С или Т>G (NARP-мутация), мутации SURF1 (недостаточность 4 комплекса), разные ядерные гены (особенно недостаточность 1 комплекса).
Лечение – витаминотерапия, L-карнитин, коэнзим Q10. Возможно использование кетогенной диеты.
