

Партнерка на США и Канаду по недвижимости, выплаты в крипто
- 30% recurring commission
- Выплаты в USDT
- Вывод каждую неделю
- Комиссия до 5 лет за каждого referral
КОМПЛЕКСНЫЕ СОЕДИНЕНИЯ, ОБРАЗУЮЩИЕСЯ В РАСТВОРЕ
ИЗ ПАРАМОЛИБДАТА АММОНИЯ, ОРТОФОСФОРНОЙ КИСЛОТЫ,
НИТРАТОВ КОБАЛЬТА ИЛИ НИКЕЛЯ И КАРБАМИДА,
И ПРИГОТОВЛЕННЫЕ НА ИХ ОСНОВЕ КАТАЛИЗАТОРЫ
ГИДРООЧИСТКИ ДИЗЕЛЬНОГО ТОПЛИВА
© 2009 г. , , , ,
Институт катализа им. СО РАН, Новосибирск
кислоту, парамолибдат аммония, нитрат кобальта или никеля и карбамид, образуется гетерополианион [Р2Мо5О23]6–, на основе которого формируются лабильные комплексы с катионами Co2+ или Ni2+. Координация катионов к гетерополианиону осуществляется через терминальные атомы кислорода в октаэдрах MoO6 и внешние атомы кислорода в группах PO4. Катализаторы, полученные нанесением этих соединений на Al2O3, проявляют высокую активность в процессе гидроочистки дизельного топлива. По этому показателю они сравнимы с лучшими зарубежными образцами и превосходят большинство отечественных промышленных катализаторов.
Для приготовления катализаторов гидроочистки широко используют нанесение соединений молибдена и кобальта или никеля на сформованный оксид алюминия [1]. Наиболее технологичным является способ однократной пропитки носителя водными растворами, содержащими одновременно молибден и кобальт (или никель). Однако при взаимодействии катионов кобальта (никеля) с молибденсодержащими анионами образуются нерастворимые соединения, например, соответствующие молибдаты. Чтобы избежать выпадения осадка, в пропиточный раствор вводят различные стабилизаторы, в том числе часто ор-тофосфорную кислоту. В зависимости от условий приготовления, концентрации и соотношения между молибденом и фосфором в растворе могут образовываться соединения с самой разной структурой. Ключевым элементом технологии приготовления катализаторов последнего поколения, предназначенных для глубокой гидроочистки нефтяных дистиллятов, является целенаправленный синтез оксидного предшественника активных центров на поверхности оксида алюминия [2]. Для этого необходимо тщательно контролировать состав и строение соединений, образующихся непосредственно в пропиточном растворе, не выделяя их в чистом виде и не применяя для их изучения рентгеноструктурные методы. Наиболее подходящей заменой является метод магнитного резонанса ядер, входящих в состав изучаемых соединений, в особенности ЯМР 17О.
Применительно к полиоксометаллатам Мо он настолько информативен, что его можно отнести к структурным методам [3, 4]. В настоящее время с помощью метода ЯМР охарактеризовано множество различных фосфор-молибденсодержащих полианионов, в том числе дифосфат-пентамолибдат [Р2Мо5О23]6– [5, 6], гетерополианионы (ГПА) Кегги-на [PMo12O40]3– [7, 8] и Доусона [P2Mo18O62]6– [5, 9], которые с высокой вероятностью образуются в пропиточных растворах в ходе приготовления катализаторов гидроочистки.
Поскольку в состав активных центров катализаторов гидроочистки – Co(Ni)MoS-фаз второго типа [10] или промотированных Ni (Co) нанокла-стеров MoS2 [11] – входит пара металлов (Co и Mo или Ni и Mo), в последнее время стали появляться публикации, посвященные изучению взаимодействия между соединениями этих металлов в пропиточных растворах [12–15]. В настоящей работе методом ЯМР на ядрах 14N, 17О, 31Р и 95Мо изучены пропиточные растворы, содержащие соединения Mo, Co или Ni, стабилизированные ортофос-форной кислотой и карбамидом. Рассмотрены взаимодействия между компонентами растворов. Катализаторы, приготовленные с использованием этих растворов, испытаны в процессе гидроочистки прямогонного дизельного топлива.
Та бл и ца 1 . Концентрации компонентов в исследованных растворах
№ раствора | Содержание компонента, моль/дм3 | ||||
H3PO4 | Мо (в составе ПМА) | Co(NO3)2 | Ni(NO3)2 | (NH2)2CO | |
1 | 0.74 | 1.47 | - | - | - |
2 | 0.74 | 1.47 | 0.15 | - | - |
3 | 0.74 | 1.47 | 0.3 | - | - |
4 | 0.74 | 1.47 | 0.74 | - | 0.75 |
5 | 0.74 | 1.47 | - | 0.15 | - |
6 | 0.74 | 1.47 | - | 0.3 | - |
7 | 0.74 | 1.47 | - | 0.74 | 0.75 |
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Приготовление растворов и катализаторов
Для изучения взаимодействий между соединениями Mo и Co (или Ni) и получения катализаторов была приготовлена серия водных растворов: для этого в воде последовательно растворяли ортофос-форную кислоту H3PO4, парамолибдат аммония (NH4)6Mo7O24 ⋅ 4H2O (далее обозначается как ПМА), нитрат кобальта Co(NO3)2 · 6H2O (Co-нитрат) или нитрат никеля Ni(NO3)2 ⋅ 6H2O (Ni-нитрат) и карбамид (NH2)2CO (табл. 1).
Катализаторы готовили на носителе Al2O3 производства ЗАО “Промышленные катализаторы” – ТНК ВР (г. Рязань) с удельной поверхностью 285 м2/г, объемом пор 0.82 см3/г и средним диаметром пор 115 Å (фракция 0.5–0.25 мм). Носитель пропитывали по влагоемкости растворами № 4 или № 7 (табл. 1), получая Co–Mo - и Ni–Mo-катализаторы соответственно. После пропитки катализаторы сушили при комнатной температуре в вытяжном шкафу в течение 12 ч и в дальнейшем использовали в процессе гидроочистки.
Образцы, прокаленные на воздухе при 550°C в течение 4 ч, содержали: Co–Mo-катализатор – 10.5% Mo, 3.21% Co и 1.3% P, Ni–Mo-катализатор – 10.6% Mo, 3.25% Ni и 1.4% P.
ЯМР-спектроскопия
Спектры ЯМР растворов с природным содержанием изотопов снимали на спектрометре “AVANCE-400 Bruker” на частотах 28.9 (14N), 54.24 (17О), 161.98 (31Р) и 26.1 (95Мо) М Гц при скорости накопления 20, 33, 0.1 и 33 Гц соответственно. Химические сдвиги (δ) измеряли относительно внешних эталонов – 4 М водного раствора Mg(NO3)2 (14N), Н2О (17О), 85%-ной Н3РО4 (31Р) и 2 М раствора Na2MoO4 (95Мо).
Испытание катализаторов в процессе гидроочистки
Катализаторы предварительно сульфидирова-ли в токе H2S (объемная скорость 500 ч–1) в течение 2 ч при атмосферном давлении и температуре 400°C. Далее катализаторы тестировали в процессе гидроочистки прямогонного дизельного топлива (содержание серы 1.05 мас. %, плотность при 20°C 0.844 г/см3, цетановое число 53.5, содержание фракции, выкипающей до 360°С, 96 об. %). Эксперименты проводили в проточной установке при давлении 3.5 МПа, весовом расходе дизельного топлива 2 ч–1 и объемном соотношении водород/дизельное топливо = 500 : 1. Гидроочистку проводили в течение суток при 340°C, затем в течение 8 ч при 370°C и снова в течение суток при 340°C. Каждые 2 ч отбирали пробы жидких продуктов, массовое содержание серы в которых определяли на рентгенофлуоресцентном анализаторе “HORIBA SLFA-20”.
РЕЗУЛЬТАТЫ И ИХ ОБСУЖДЕНИЕ
Параметры спектров ЯМР приготовленных растворов приведены в табл. 2. При изучении взаимодействия между компонентами пропиточных растворов необходимо учитывать не только положение и интенсивность пиков, но и их внешний вид. Наиболее информативные фрагменты полученных спектров приведены на рис. 1–3. Растворы характеризуются величинами pH от 2.5 до 3.5, молибден находится в них в виде полиядерных соединений – полиоксомолибдатов. В исходном растворе № 1 мольное отношение Мо/H3PO4 = 2 : 1. Вид спектра ЯМР 31Р (рис. 1) показывает, что бóльшая часть фосфора находится в составе комплекса с молибденом в форме соединения с δ = = 1.94 м. д., а меньшая – в виде свободного фосфата (δ = 0.87 м. д.). Сопоставление интенсивно-стей пиков и концентраций компонентов в растворе (0.6 моль/дм3 H3PO4 находится в закомплексованной форме и 0.14 моль/дм3 – в свободной)
Та б л и ц а 2 . Параметры спектров ЯМР растворов № 1–7
М2+/Мо (ат.) | Параметры спектров ЯМР на ядрах | ||||||||||
95Мо (при 294 К) | 31P (при 294 К) | 17O (при 323 К) | |||||||||
раствора | 8, м. д. | ширина пика, Гц | 8, м. д. | ширина пика, Гц | О=Mo | Mo–О–Mo | PO4 | ||||
8, м. д. | ширина пика, Гц | 8, м. д. | ширина пика, Гц | 8, м. д. | ширина пика, Гц | ||||||
1 2 3 4 5 6 7 | Со/Мо = 0 Со/Мо= 0.1 Со/Мо= 0.2 Со/Мо= 0.5 Ni/Мо = 0.1 Ni/Мо = 0.2 Ni/Мо = 0.5 | 12.6 + 1 60 + 2 180 + 3 16.4 + 2 16.6 + 2 15.4 + 3 | 260 550 830 570 680 820 | 1.9 13 3.7 6 9.3 | 90 3700 180 310 650 | 837 935 839 851 860 | 160 1500 700 1100 400 | 379 380 385 390 381 383 386 | 320 320 380 540 220 370 220 | 86 97 137 168 90 | 490 600 870 1500 300 |
* Сигнал не зарегистрирован из-за большой ширины пика.
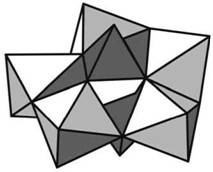
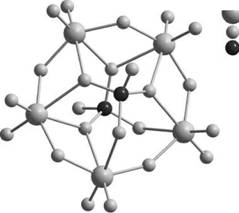
свидетельствует, что в полученном комплексном соединении на 5 атомов молибдена приходится 2 атома фосфора. В спектре ЯМР 17О раствора № 1 (рис. 2) имеются линии, отнесенные к атомам кислорода разного типа, связанным с молибденом: терминальным О=Мо (δ = 837 м. д.), мости-ковым Mo–O–Mo (δ = 379 м. д.) и атомам кислорода в составе фосфата РО4. Широкий пик с δ = = 86 м. д. (табл. 1) появляется в результате наложения линий от иона PO34 -, как свободного, так и входящего в состав комплексного соединения с молибденом. В спектре ЯМР 95Mo имеется только одна линия (табл. 2), и это указывает, что в растворе № 1 весь молибден находится в составе одного соединения. Соотношение интенсивностей концевых и мостиковых атомов О, связанных с молибденом, а также химические сдвиги в спектрах ЯМР 31Р и 95Мо хорошо согласуются данными [6], где методом ЯМР в растворе был изучен дифосфат-пентамолибдат [Р2Мо5О23]6–. Значит, в растворе № 1 весь молибден входит в состав ГПА [Р2Мо5О23]6–, а избыточный фосфор, не входящий в состав этого аниона, находится в виде свободного фосфата. Строение [Р2Мо5О23]6–, отвечающее структурным данным [16], показано на рис. 3. Ди-фосфат-пентамолибдат имеет вид плоского кольца, образованного пятью октаэдрами MoO6, которые соединяются между собой пятью кислородными мостиками, а также двумя фрагментами РО4, расположенными над и под плоскостью кольца. Таким образом, в ходе приготовления пропиточного раствора с указанными выше концентрациями компонентов на первой стадии из ПМА и H3PO4 селективно образуется ГПА [Р2Мо5О23]6–, аналогично тому, как это происходит при взаимодействии ПМА с фосфорной кислотой, адсорбированной на поверхности Al2O3 [17].
В последние годы различные молибденсодержа-щие гетерополисоединения широко используют для приготовления высокоактивных катализаторов гидроочистки [18, 19]. Однако практически во всех известных ГПА, содержащих молибден и кобальт (или никель), атомное отношение Co(Ni)/Mo значительно ниже, чем в катализаторах гидроочистки, где оно близко к 0.5 [14, 19–23]. Необходимого для катализа соотношения металлов можно достичь, заменив катионы NH4+ в гетерополисоединениях на Co2+ или Ni2+. Поэтому разработаны специальные методики обмена катионов аммония на катионы кобальта или никеля [24, 25]. При этом количество катионов Co2+, взаимодействующих с ГПА, определяется его зарядом. Возможны два типа взаимодействия.
1) Образование комплексных соединений, в которых какие-либо атомы кислорода входят одновременно и в состав октаэдров MoO6, и в бли
жайшее кислородное окружение кобальта (никеля). В этом случае ГПА является макролигандом, координированным к Со2+ (или Ni2+), аналогично координации [NiMo9O32]6– к катионам La3+ и Pr3+ [26], а также координации ГПА [ZMo12O42]8– (где Z = Ce, Th, U) к катионам редкоземельных элементов [27] или Cu2+ [28]. Как правило, координация катионов к ГПА осуществляется через концевые (терминальные) атомы кислорода [12, 19, 24, 26–28].
2) Образование ионных пар между ГПА и аква-ионами кобальта (никеля), подобных описанным в публикациях [19, 26].
При определенных условиях (определенных концентрациях компонентов раствора, природе и заряде катиона и ГПА) могут реализовываться взаимодействия обоих типов. Так, анион [Co2Mo10O38H4]6– способен взаимодействовать с тремя катионами Co2+, образуя биметаллическое соединение
[Co2Mo10H4O38{Co(H2O)5}2]2– · (Co(H2O)6)2+ · (H2O)9;
при этом два катиона Co2+ координированы к ГПА через терминальные атомы кислорода, а один присутствует в растворе или в кристаллах в
виде акваиона Co(H2O)62 +, который связан с ГПА ионными силами [19].
Для изучения способа координации катионов кобальта или никеля по отношению к аниону [Р2Мо5О23]6– были приготовлены растворы с различными концентрациями Co2+ (Ni2+) (табл. 1). Известно, что при образовании комплексов ГПА с катионами различных металлов в спектрах ЯМР растворов происходит уширение и смещение сигналов 95Mo и 17O, которое усиливается с ростом концентрации катионов [4, 26–29]. Благодаря парамагнетизму катионов Co2+, Cu2+ или Ni2+ в случае их координации к ГПА наблюдается более значительное уширение и смещение сигнала. Как правило, в спектрах ЯМР 17O более сильные изменения характерны для сигналов от тех атомов кислорода, которые непосредственно участвуют в комплексообразовании.
|
 (a)
(a)
Рис. 3. Строение аниона [Р2Мо5О23]6–: а – атомная модель, б – модель в виде октаэдров и тетраэдров.
В соответствии с этими наблюдениями изменения во всех изученных нами спектрах усиливаются с ростом отношения Co/Mo в растворе (табл. 2). В спектрах ЯМР 95Mo отмечено монотонное уширение пика, сопровождающееся парамагнитным сдвигом в область слабого поля, при этом линия 95Mo всегда остается одиночной. В спектрах ЯМР 31P происходят аналогичные изменения. В спектрах ЯМР 17O (рис. 2, табл. 2) сигналы терминальных атомов кислорода Mo=O и атомов кислорода, входящих в состав фрагмента PO4, с ростом концентрации кобальта в растворе сильно уширяются и смещаются в область слабого поля, в то время как изменения пика мостико-вого кислорода Mo–O–Mo невелики. Присутствующий в спектрах 17O узкий сигнал от свободного аниона NO3-, вводимого в раствор вместе с кобальтом, смещается в направлении слабого поля относительно его положения в растворах диамагнитных солей. При добавлении в раствор вместо Со2+ ионов Ni2+ наблюдаются аналогичные изменения, но из-за меньшего магнитного момента никеля они при той же концентрации парамагнитного иона меньше (табл. 2).
Таким образом, особенности изменений в спектрах ЯМР 95Mo,31P и 17O раствора [Р2Мо5О23]6– при добавлении к нему различных количеств катионов Co2+ или Ni2+ однозначно указывают на образование лабильных комплексов ГПА–парамаг-нитный ион. При этом координация парамагнитных катионов к ГПА осуществляется через терминальные атомы кислорода Mo=O и внешние атомы кислорода в фрагменте PO4. В соответствии со структурой ГПА (рис. 3) наиболее вероятным представляется расположение двух катионов Co2+ над и под плоскостью пятичленного кольца, причем в координационную сферу каждого катиона кобальта может входить по одному терминальному атому кислорода из октаэдра MoO6 и по одному внешнему атому кислорода из фрагмента PO4. В растворах № 4 и № 7, предназначенных для приготовления катализаторов, на один анион [Р2Мо5О23]6– приходится по 2.5 катиона кобальта или никеля. При осаждении связь между избыточными катионами кобальта (никеля) и ГПА может осуществляться благодаря ионному взаимодействию, подобно тому, как это происходит в соединении [Co2Mo10H4O38(Co(H2O)5)2]2– · (Co(H2O)6) · · (H2O)9 [19].
Поскольку для приготовления катализаторов гидроочистки применяют растворы с высокой концентрацией молибдена и кобальта (никеля), при их длительном хранении происходит нежела-тельное выпадение кристаллического осадка. Этого можно избежать, введя в раствор стабилизирующий агент – карбамид. Очевидно, карбамид образует с другими компонентами раствора хорошо растворимые комплексные соединения, что и было подтверждено методом ЯМР 14N (рис. 4). В спектре ЯМР 14N присутствуют линии аниона NO3 (5 = –2 м. д.), вводимого в раствор вместе с Со2+, и иона аммония NH4 (5 = –360 м. д.), а также один уширенный сигнал карбамида, смещенный относительно линии свободного карбамида на 24 м. д. в сторону слабого поля. Такое уширение и смещение сигнала карбамида указывают на его координацию к парамагнитным катионам кобальта через атомы азота и на лабильность комплекса. Для раствора № 7, содержащего катионы Ni2+, отмечен аналогичный характер взаимодействия компонентов.
Таким образом, из сопоставления данных ЯМР 14N, 17O, 31Р и 95Мо следует, что при приготовлении катализаторов гидроочистки с использованием парамолибдата аммония, ортофосфорной кислоты, нитрата кобальта или никеля и карбамида в пропиточном растворе образуется биметаллическое соединение, представляющее собой ГПА [Р2Мо5О23]6–, к которому через атомы кислорода – терминальные и внешние атомы О в PO4 – координированы катионы Co2+ или Ni2+, стабилизируемые в растворе карбамидом.
Если катализаторы готовят без высокотемпературного прокаливания, структура образующихся в растворе в результате координации кобальта к молибденсодержащим анионам биметаллических соединений не изменяется при нанесении их на поверхность носителя [12, 14, 19, 20]. Такие соединения при последующем сульфидировании селективно превращаются в активные центры гидрогенолиза C–S-связи, чем предопределяется высокая активность полученных катализаторов в реакциях гидроочистки. Катализаторы, приготовленные в настоящей работе пропиткой Al2O3 растворами № 4 и № 7, перед сульфидированием высушивали при комнатной температуре, что не должно было привести к разложению полученных биметаллических соединений.
Результаты испытания Co–Mo - и Ni–Mo-ката-лизаторов в процессе гидроочистки приведены на рис. 5. На обоих катализаторах конверсия серосодержащих соединений превышала 99%. Co–Mo-катализатор проявил несколько более высокую гидрообессеривающую активность, однако и на Ni–Mo-образце остаточное содержание серы при 340°C было очень низким и не превышало 60 ppm, а при 370°C оно понижалось до 20–30 ppm. Оба катализатора весьма устойчивы к дезактивации, о чем свидетельствует лишь незначительное повышение остаточного содержания серы при возвращении к температуре 340°С после 8 ч работы при 370°C (рис. 5).
Особый интерес представляло сравнение каталитических свойств исследованных нами и наиболее известных отечественных и импортных нанесенных катализаторов гидроочистки. Поскольку у нас не было официального разрешения на тестирование промышленных катализаторов, в особенности зарубежных, мы сопоставляли наши результаты с данными, опубликованными в открытой печати. С этой целью наши сульфидиро-ванные катализаторы были испытаны в процессе гидроочистки прямогонного дизельного топлива в следующих условиях: весовой расход сырья 2 ч–1, давление 3.5 МПа, объемное соотношение водород/сырье = 500 нм3/м3, температура 340 и 370°C. Эти показатели находятся в пределах тех параметров, которые обычно используют зарубежные [10, 30–33] и российские [34–38] исследователи при
тестировании катализаторов глубокой гидроочистки дизельного топлива как в лабораторном, так и в укрупненном масштабе: весовой расход топлива 1.2–3.0 ч–1, давление 3–4.5 МПа, температура 340–380°C, объемное соотношение H2/сырье от 250 до 500 нм3/м3. Как правило, получаемое в этих условиях на лучших зарубежных катализаторах (KF-757 Stars, TK-554, TK-574, TK-576 BRIM) дизельное топливо содержит от 10 до 100 ppm остаточной серы. На распространенных отечественных катализаторах результаты значительно хуже [38]. Так, на катализаторах РК-231 и РК-242 остаточное содержание серы составляет 100–350 ppm [36], на НКЮ-232 и НКЮ-500 – в среднем 350– 500 ppm [37], а на системах ГКД-205 и ГКД-300 – не ниже 300 ppm при 380°C [35].
В целом описанные в настоящей работе катализаторы по активности сопоставимы с лучшими зарубежными образцами и значительно превосходят распространенные отечественные катализаторы. При близких условиях проведения процесса гидроочистки на них достигается значительно более низкое содержание остаточной серы в дизельном топливе.
Таким образом, установлено, что при синтезе катализаторов гидроочистки из парамолибдата аммония, ортофосфорной кислоты, нитрата кобальта или никеля и карбамида в пропиточном растворе образуется ГПА [Р2Мо5О23]6–, к которому через терминальные атомы кислорода в октаэдрах MoO6 и внешние атомы кислорода в группе PO4 координированы катионы Co2+ или Ni2+, так что создается лабильный комплекс. Стабилизация раствора обеспечивается введением карбамида, который координируется через атомы азота к ионам Co2+ (Ni2+). Катализаторы, полученные нанесением указанных соединений на Al2O3, проявляют высокую активность в процессе гидроочистки дизельного топлива, они сравнимы с лучшими зарубежными образцами и превосходят распространенные отечественные промышленные катализаторы.
СПИСОК ЛИТЕРАТУРЫ
1. , , Катализаторы процессов углубленной переработки нефти. М.: Химия, 1992. 272 с.
2. , , и др. // Мат. 7-го Межд. форума “Топливно-энергетический комплекс России: региональные аспекты”. СПб: Рестэк, 2007. С. 245.
3. , // Журн. структ. химии. 2006. Т. 47. С. 961.
4. , , Ядерный магнитный резонанс в неорганической химии. М.: Наука, 1988. 216 с.
5. , , // Изв. АН СССР. Сер. хим. 1980. № 3. С. 709.
6. , , // Изв. АН СССР. Сер. хим. 1980. № 7. С. 1477.
7. Filowitz M., Ho R. K., Klemperer W. G., Shum W. // In-org. Chem. 1979. V. 18. P. 93.
8. , , и др. // Докл. АН СССР. 1978. Т. 240. С. 117.
9. , // Журн. неорг. химии. 1995. Т. 40. С. 1369.
10. Eijsbouts S., Van den Oetelaar L. C.A., Van Ruijenbroek R. R. // J. Catal. 2005. V. 229. P. 352.
11. Lauritsen J. V., Kibsgaard J., Olesen G. H. et al. //J .Cat-al. 2007. V. 249. P. 220.
12. Bergwerff J. A., Visser T., Weckhuysen B. M. // Catal. Today. 2008. V. 130. P. 117.
13. Van Dillen A. J., Terorde R. J.A. M., Lensveld D. J. et al. // J. Catal. 2003. V. 216. P. 257.
14. Blanchard P., Lamoier C., Griboval A., Payen E. // Ap-pl. Catal. A: General. 2007. V. 322. P. 33.
15. Van de Water L. G.A., Bergwerff J. A., Leliveld B.(R.)G. et al. // J. Phys. Chem. B. 2005. V. 109. P. 14513.
16. Strandberg R. // Acta Chem. Scand. 1973. V. 27. P. 10 04 .
17.Van Veen J. A.R., Hendriks P. A.J. M., Andrea R. R. et al. // J. Phys. Chem. 1990. V. 94. P. 5282.
17. , , // Рос. хим. журн. (Журн. Рос. хим. об-ва им. леева). 2008. Т. 52. № 4. C.41.
18. Martin C., Lamonier C., Fournier M. et al. // Chem. Mater. 2005. V. 17. P. 4438.
19. Mazurelle J., Lamonier C., Lancelot C. et al. // Catal. Today. 2008. V. 130. P. 41.
20. Chianelli R. R., Daage M., Ledoux M. J. // Adv. Catal. 1994. V. 40. P. 177.
21. Pettiti I., Botto I. L., Cabello C. I. et al. // Appl. Catal. A: General. 2001. V. 220. P. 113.
22. Сульфидные катализаторы: синтез, структура, свойства. Новосибирск: Новосибирское акад. изд-во “Гео”, 2007. С. 206.
23. Martin C., Lamonier C., Fournier M. et al. // Inorg. Chem. 2004. V. 43. P. 4636.
24. Griboval A., Blanchard P., Payen E. et al. // rf. Sci. Catal. 1997. V. 106. P. 181.
25. , , Тор-ченкова Е. А. // Журн. неорг. химии. 1990. Т. 35. С. 2100.
26. , , // Журн. неорг. химии. 1990. Т. 35. С. 2104.
27. , , // Журн. неорг. химии. 1990. Т. 35. С. 2110.
28. , , // Журн. неорг. химии. 1989. Т. 34. С. 849.
29. Topsøe H., Hinnemann B., Norskov J. K. et al. // Catal. Today. 2005. V. 107–108. P. 12.
30. Knudsen K. G., Cooper B. H., Topsшe H. // Appl. Catal. A: General. 1999. V. 189. P. 205.
31. EPA-Diesel RIA, Regulatory Impact Analysis: Heavy-Duty Engine and Vehicle Standards and Highway Diesel Fuel Sulfur Control Requirements, United States Environmental Protection Agency, Air and Radiation, EPA420-R-00-026. December 2000.
32. Frizi N., Blanchard P., Payen E. et al. // Catal. Today. 2008. V. 130. P. 32.
33. , , и др. // Химия и технология топлив и масел. 2007. № 1. С. 3.
34. , , и др. // Химия и технология топлив и масел. 2007. № 1. С. 10.
35. , , и др. // Нефтепереработка и нефтехимия. 2007. № 6. С. 13.
36. , , и др. // Катализ в пром-сти. 2004. № 1. С. 25.
37. , , и др. // Катализ в пром-сти. 2008. Спецвыпуск. С. 6.
