
Партнерка на США и Канаду по недвижимости, выплаты в крипто
- 30% recurring commission
- Выплаты в USDT
- Вывод каждую неделю
- Комиссия до 5 лет за каждого referral
Московский Государственный Университет
им.
Факультет биоинженерии и биоинформатики
Молекулярная динамика комплекса тетрациклина с 30S рибосомной субчастицой
Курсовая работа
Научные руководители:
М. н.с. к. х.н.
Москва
2003 г.
Введение
В настоящее время антибиотики являются основным средством борьбы с бактериальными инфекциями человека и животных. Тетрациклин (Тс) является традиционным популярным антибиотиком широкого спектра действия, который используется как в медицине, так и ветеринарии.
Тетрациклиновые антибиотики привлекли большое внимание исследователей благодаря их важному практическому значению. Обладая широким антибиотическим спектром (активны в отношении грамм-положительных и грамм-орицательных бактерий, риккетсий и др.), эти соединения нашли широкое применение в медицинской практике и ветеринарии. Предполагается, что он проникает в бактериальную клетку путем пассивного транспорта через мембрану и взаимодействует с рибосомой, ингибируя трансляцию, что приводит к гибели клетки. В клетках прокариот основной мишенью для Тс является рибосома, взаимодействие с которой приводит к ингибированию биосинтеза белка.
На сегодня существуют различные данные о положении тетрациклина в составе рибосомы. В работе Yonath [1] тетрациклин связывается с малой субчастицей рибосомы в 6 местах, и какое место важно, до сих пор не ясно. Так как полученные Yonath [1] методом РСА данные очевидно избыточны в данной работе, предпринята попытка оценить структурную специфичность мест связывания Тс. Для решения этой задачи нами был использован метод молекулярной динамики (МД).
Метод молекулярной динамики
В методе молекулярной динамики рассчитывается движение атомов в молекулярной системе с заданными взаимодействиями между ними путем численного интегрирования уравнений движения частиц.
Основу метода составляет численное решение системы ![]() дифференциальных уравнений второго порядка вида:
дифференциальных уравнений второго порядка вида:
![]()
где![]() – масса
– масса 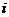 -того атома,
-того атома, ![]() - радиус вектор,
- радиус вектор,![]() - суммарная сила, действующая на
- суммарная сила, действующая на ![]() -ый атом со стороны всей моделируемой системы, а
-ый атом со стороны всей моделируемой системы, а ![]() – число частиц в системе.
– число частиц в системе.
Сила ![]() задается как частная производная потенциальной энергии системы
задается как частная производная потенциальной энергии системы ![]() по координате -той частицы:
по координате -той частицы:
![]()
![]() зависит от расположения всех частиц в системе и от дополнительных внешних сил, участвующих в расчете. Таким образом, сила
зависит от расположения всех частиц в системе и от дополнительных внешних сил, участвующих в расчете. Таким образом, сила ![]() является потенциальной и зависит только от внутренней геометрии системы, но не зависит от положения центра масс системы.
является потенциальной и зависит только от внутренней геометрии системы, но не зависит от положения центра масс системы.
Силовое поле
Набор аналитических зависимостей соответствующих компонент потенциальной энергии в совокупности с набором параметров для этих зависимостей называют си полем. Существуют несколько групп силовых полей: AMBER, CHARMM, GROMOS и другие [2,3]. Все из указанных силовых полей имеют как общие аналитические зависимости для потенциальных энергий различных типов взаимодействий, так и оригинальные. В связи с этим силовые поля несовместимы друг с другом по силовым постоянным. Следует также отметить, что при создании каждого силового поля многие константы определяются методом оптимизации путем численного подбора значений физических величин для наилучшего соответствия данных расчетных и экспериментальных данных. Поэтому любое силовое поле является самосогласованным и не может использоваться по частям. В данной работе в качестве силового поля было выбрано поле GROMACS(v.3.1.4) [3].
Материалы и методы
1. Построение непрерывного окружения
Так как метод МД накладывает ограничения на размер системы (количество атомов), мы решили экстрагировать сайт связывания тетрациклина №4. Выбор именно этого сайта обусловлен тем, что этот сайт был прежде не описан в литературе и при этом согласно результатам анализа (работа студента ) консервативности сайтов связывания он является самым консервативным.
Мы пытались минимизировать количество разрывов в цепи рРНК окружения четвертого тетрациклина. Для получения этой структуры использовали программу Swiss pdb viewer [4]. В этой программе открыли файл 30S субчастицы рибосомы с шестью тетрациклинами. Сначала выключили все и оставили только четвертый тетрациклин, чтобы исключить работу со всей 30S субчастицей. Затем выделили всех соседей на расстоянии 16А для того, чтобы построить окружение с минимальным количеством разрывов цепи РНК. На расстоянии меньше 16А у нас выделялись лишь обрывки цепи РНК. После чего удалили те участки, которые не явно взаимодействуют с тетрациклином (отдельные куски РНК, не влияющие на построение окружения). Далее построили сплошную молекулярную поверхность и проследили за тем, чтобы тетрациклин не “вылезал из нее”, т. е. чтобы площадь Тс, доступная солвенту, не изменялась. Затем проделали то же самое, но уже в целой субчастице. Результаты оказались одинаковыми. У нас получилось четыре цепи и порядка 60 нуклеотидов, образующих непрерывное окружение.
2. Получение файлов координат и топологии.
Все операции проводились в операционной системе Irix. Молекулярное моделирование, минимизация энергии, динамические расчеты и анализ полученных траекторий проводились с использованием пакета программ GROMACS. Для расчета был написан управляющий batch файл, содержащий в себе последовательность исполняемых команд, которые представляют собой расчетные модули программного пакета GROMACS 3.0. Содержание файла представлено ниже:
 вакууме
grompp -f em -c ${MOL}.gro -p ${MOL} -o ${MOL}_em
grompp для добавления ионов натрия
grompp -f em -c ${MOL}_b4em.gro -p ${MOL} -o ${MOL}_em
#меняет воду на натрий
genion -np 133 -s ${MOL}_em.tpr -o ${MOL}_b4em3.gro
#минимизация энергии с натрием
grompp -f em -c ${MOL}_b4em3 -p ${MOL} -o ${MOL}_em
mdrun -nice 4 -s ${MOL}_em -o ${MOL}_em -c ${MOL}_b4pr -v
#posission restaing(утряска воды)
grompp -n -f pr -c ${MOL}_b4pr -r ${MOL}_b4pr -p ${MOL} -o ${MOL}_pr
mdrun -nice 4 -s ${MOL}_pr -o ${MOL}_pr -c ${MOL}_b4md -v
#молекулярная динамика
grompp -f md -c ${MOL}_b4md -p ${MOL} -o ${MOL}_md
mdrun -nice 4 -s ${MOL}_md -o ${MOL}_md -c ${MOL}_after_md -v &
#переделывает файл траектории в pdb
#trjconv -b 1500 -e 2500 -skip 10 -f ${MOL}_md -s ${MOL}_md -o ${MOL}_md_out10.pdb
" width="697 height=414" height="414""/>
вакууме
grompp -f em -c ${MOL}.gro -p ${MOL} -o ${MOL}_em
grompp для добавления ионов натрия
grompp -f em -c ${MOL}_b4em.gro -p ${MOL} -o ${MOL}_em
#меняет воду на натрий
genion -np 133 -s ${MOL}_em.tpr -o ${MOL}_b4em3.gro
#минимизация энергии с натрием
grompp -f em -c ${MOL}_b4em3 -p ${MOL} -o ${MOL}_em
mdrun -nice 4 -s ${MOL}_em -o ${MOL}_em -c ${MOL}_b4pr -v
#posission restaing(утряска воды)
grompp -n -f pr -c ${MOL}_b4pr -r ${MOL}_b4pr -p ${MOL} -o ${MOL}_pr
mdrun -nice 4 -s ${MOL}_pr -o ${MOL}_pr -c ${MOL}_b4md -v
#молекулярная динамика
grompp -f md -c ${MOL}_b4md -p ${MOL} -o ${MOL}_md
mdrun -nice 4 -s ${MOL}_md -o ${MOL}_md -c ${MOL}_after_md -v &
#переделывает файл траектории в pdb
#trjconv -b 1500 -e 2500 -skip 10 -f ${MOL}_md -s ${MOL}_md -o ${MOL}_md_out10.pdb
" width="697 height=414" height="414""/>
Программа pdb2gmx переводит исходный .pdb фаил в рабочий формат .top файлов топологии GROMACS. В связи с тем, что Gromacs понимает лишь стандартные молекулы (РНК, ДНК, белок), мы удалили из pdb файла тетрациклин. После чего остатки в pdb были переименованы, а сами координаты четвертого тетрациклина были отправлены на сервер (The Dundee PRODRG Server, http://davapc1.bioch. dundee. ac. uk/programs/prodrg/prodrg. html )[5] для получения файлов координат и топологии. На выдаче сервера были получены координаты (файл. gro) и файл топологии (файл. top) для четвертого тетрациклина. С помощью программы pdb2gmx были получены gro и top файлы для РНК. После чего в файл gro для РНК был добавлен файл gro тетрациклина, в файл top для РНК был добавлен файл top для тетрациклина.
3. Создание физиологического раствора
Подправили ячейку до ее приемлемого размера (до отступа от края молекулы 2.0 нм) с помощью программы editconf. Затем сгенерировали воду в ячейке с помощью программы genbox. После чего запустили препроцесс непосредственно МД, с помощью программы grompp для того, чтобы программа genion получила файл для добавления ионов. Запустили genion (заменяет молекулы воды на ионы Na+ в тех местах, где необходим положительный заряд). Затем запустили препроцесс (программа grompp) и для минимизации энергии, после чего запустили собственно минимизацию энергии (программа mdrun). Метод молекулярной динамики может не сработать, если начальная конфигурация системы далека от равновесия и внутренние силы слишком велики. В этих случаях требуется минимизация энергии системы. Другая причина для выполнения минимизации энергии – это устранение всей кинетической энергии из системы. Это бывает необходимо при сравнении нескольких мгновенных состояний системы при динамическом моделировании, так как минимизация энергии устраняет тепловой шум в структурах и в потенциальных энергиях, и, следовательно, качество сравнения повышается. Параметры для минимизации энергии содержатся в файле em. mdp. Провели МД по position restraint (утрясли воду в ячейке) . Параметры для position restraint(для утряски воды в ячейке) содержатся в файле pr. mdp. Запустили собственно динамику с помощью программы mdrun. mdrun — интегратор GROMACS, рассчитывает траекторию системы с заданным в файле .mdp временным шагом, температурой, давлением и другими условиями. Использует входной файл .tpr, генерирует выходной файл .trr, или его сокращенный вариант .xtc, содержащий в себе запись состояний системы (координаты и скорости атомов, энергию системы, силы) через заданные интервалы времени.
4. Анализ полученных данных
С помощью программы g-sas(рассчитывает поверхность, доступную растворителю в каждый момент времени)был получен файл area. xvg, с помощью программы g_dist(считает расстояние между центрами масс двух групп атомов как функцию от времени)получили файл dist. xvg(для получения этого файла в файле index. ndx, который подается команде g_dist на вход, пришлось отметить нуклеотид CYT1323,от которого происходил расчет, и TET4, до которого происходил расчет), с помощью программы g_msd(рассчитывает в каждый момент времени среднеквадратичное отклонение для атомов в группе от начального состояния)получили файл msd. xvg, с помощью программы g_energy(рассчитывает общую энергию системы в каждый момент времени) получили файл energy. xvg.
Результаты
Движение тетрациклина в процессе динамики было визуализировано с помощью программы PyMOL [6].
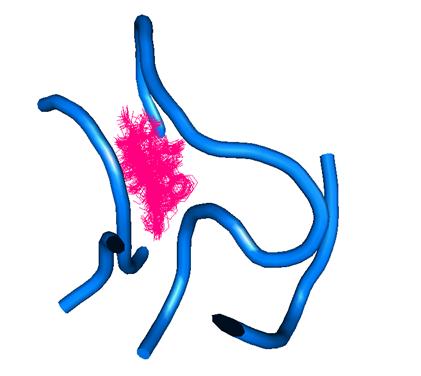
Рис.1. Движение Тс в Тет4 учаске
На основании полученных данных был проведён анализ изменения параметров динамики:
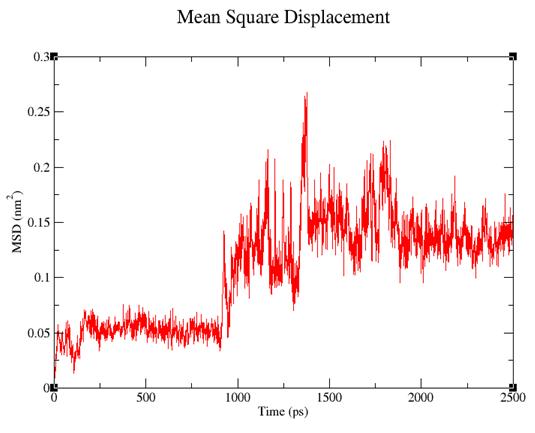
Рис.2. Зависимость отклонения атомов от первоначального состояния во времени.
Рис. 3. Изменение общей энергии системы во времени.
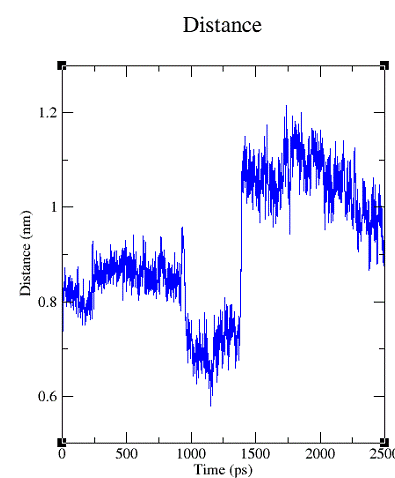
Рис.4.Изменение значений расстояний (в нанометрах) между центрами масс тетрациклина и CYT1323 (нуклеотид с ближайшим центром масс) во времени.
При анализе отклонений было выявлено, что в начале тетрациклин колеблется незначительно, затем, начиная примерно с 1000-ой пикосекунды, подвижность Тс увеличивается, а затем снова падает. Это означает, что Тс занял другую позицию.
Из графика, отражающего изменение дистанций между атомами видно, что расстояния сначала уменьшаются, затем увеличиваются. Это свидетельствует о том, что тетрациклин отдалился от находящегося с ним в близком контакте CYT1323 (нуклеотид с ближайшим центром масс).Но, опять-таки, отклонение незначительно.
В ходе динамики общая энергия системы падает. Это позволяет предположить, что в ходе симуляции молекулярной динамики Тс занимает более энергетически выгодную позицию.
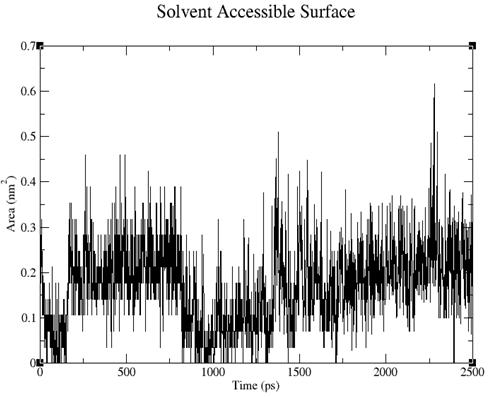
Рис. 5. Изменение площади поверхности тетрациклина, доступной растворителю, во времени.
Площадь поверхности отображает взаимодействие тетрациклина с окружающей его РНК. Сначала площадь уменьшилась, следовательно, взаимодействие увеличилось. Затем поверхность периодически то увеличивалась, то уменьшалась, что свидетельствует о динамическом колебании количества контактов тетрациклина с РНК. В результате тетрациклин занял более выгодное положение.
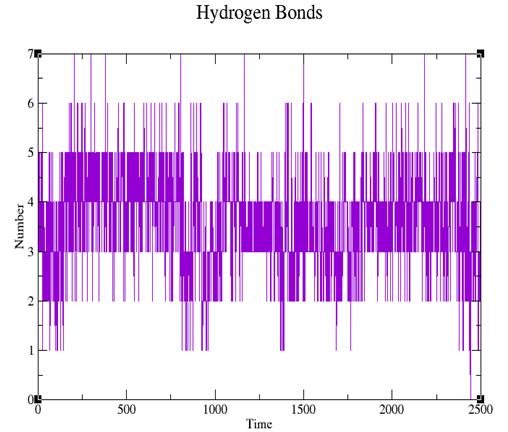
Рис. 6. Изменение количесва водородных связей, образуемых тетрациклином, во времени.
Из графика видно, что количество водородных связей остается в среднем неизменным. Вероятно, что движущей силой движения тетрациклина в ходе нашей симуляции является оптимизация гидрофобных взаимодействий.
Выводы
· Методом МД проведена минимизация общей энергии системы с –79095 до –143744 кДж/моль. Молекула тетрациклина сместилась на 3.87 А.
· Методом МД показано, что положение тетрациклина в месте связывания №4 стабильно.
Список использованной литературы
[1] Pioletti, M. et al. (2001) Embo J 20, 1829-39.
[2] Berendsen, H. J.C., van der Spoel, D., van Drunen, R. (1995) Comp. m. 91, 43–56.
[3] Lindahl, E., Hess, B., van der Spoel, D. (2001) J. Mol. Mod 7, 306–317.
[4] Schwede, T., Kopp, J., Guex, N. and Peitsch, M. C. (2003) Nucleic Acids Res 31, 3381-5.
[5] D. M.F. van Aalten, R. B., J. B.C. Findlay, M. Hendlich, R. W.W. Hooft and G. Vriend (1996) Journal of Computer Aided Molecular Design 10, 255-262.
[6] DeLano, W. L. (2002) on World Wide Web http://www. pymol. org.
