

Партнерка на США и Канаду по недвижимости, выплаты в крипто
- 30% recurring commission
- Выплаты в USDT
- Вывод каждую неделю
- Комиссия до 5 лет за каждого referral
Московский Государственный Университет им.
Химический факультет
Кафедра аналитической химии
Зачетная задача по аналитической химии
Выполнил:
стулент 211 группы
Преподаватель:
к. х.н. доцент
Москва 2009 г.
Оглавление
1.Описание объекта
2.Качественный анализ
3.Выбор методики определения меди
3.1.Краткая характеристика методик определения меди
3.1.1.Титриметрическое определение
3.1.2.Гравиметрическое определение
3.2.Обсуждение выбора методики определения меди
3.3.Обоснование выбора методики
4.Определение меди в образце
4.1.Схема количественного анализа образца
4.2.Расчет навески
4.3.Растворение пробы
4.4.Методики определения
4.4.1.Иодометрическое определение
4.4.2.Комплексонометрическое определение
4.4.3Приготовление и подготовка реактивов
4.5.Результаты определения
4.6.Описание особенностей или сложностей анализа
4.7.Выводы
5.Список литературы
1.Описание объекта
Образец представляет собой стружки желтого цвета, равномерно окрашенные, с металлическим блеском, достаточно тяжелые. Можно предположить, что это сплав, основным компонентом которого является медь.
2.Качественный анализ
Схема качественного анализа образца, включающая предварительные испытания, приведена в табл.1.
Таблица 1 (схема качественно анализа сплава)
№ | образец | Реагент, условия | Наблюдения | Предварительные Выводы | Предполагаемый состав | |
осадок | раствор | |||||
1 | Исходный | HNO3 6М | полное растворение, цвет раствора – голубой | Cu, отс. Sn, Sb в знач. кол-вах | Cu2+ | |
2 | Раствор 1 | NH3 (25%) | ярко-синее окрашивание раствора | Cu | ||
3 | Раствор 1 | CH3COOH(тв.) до рН 5 (NH4)Hg(SCN)4 | фиолетовые крестовидные кристаллы | Zn | ||
4 | Раствор 1 | H2SO4(1:1), t°, до белых паров, охладить, растворить в H2O | Раствор голубой, осадка нет | отс. Pb в знач. кол-вах | ||
5 | Раствор 4 | NH4Cl(раствор) | осадка нет | отс. Al в знач. кол-вах | ||
6 | Раствор 4 | Диметилглиоксим, NH3, на фильтр. бумагу (свидетель) | Цвет бумаги зеленый, исчезает после р-рения в H2O(свидетель) | отс. Ni | ||
7 | Раствор 4 | Na2S2O3 | Выпадение осадка оранжевого цвета | Cu2S | ||
8 | Раствор 7 | ПАН, экстракция в CCl4(свидетель) | Фиолетовое окр. обеих фаз | отс. Mn | ||
9 | Раствор 4 | Na3BiO3, HNO3(конц.) | отс. малиновой окраски | отс. Mn | ||
10 | Раствор 7 | HCl до рН~1, дитизон в CCl4(экстракция Cu), 3к гидротартрата Na, 2к диметилглиоксима, NaOH 30% до рН~10, 3к дитизона, CCl4 | орг. фаза красно-бурая | Cd2+ в следовых кол-вах |
Вывод: образец содержит медь и цинк в качестве основных компонентов, а также Cd в следовых количествах.
Для количественного определения выбрана медь.
3.Выбор методики определения меди
3.1.Краткая характеристика методик определения меди
3.1.1.Титриметрическое определение
Комплексонометрическое титрование[1]
Прямое титрование с ЭДТА протекает по уравнению
![]()
Титрование возможно в широком интервале рН (3-11), выбор индикатора зависит от значения рН. В слабокислых средах (ацетатный буфер) чаще всего используют ПАН (1-(2-пиридилазо)-2-нафтол) или ПАР (4-(2-пиридилазо)резорцин), в слабощелочных (аммиачный буфер) – мурексид. Присутствующий в сплаве цинк также реагирует с ЭДТА в выбранных условиях. Для определения меди в присутствии цинка сначала оттитровывают сумму Cu и Zn, затем маскируют медь тиосульфатом 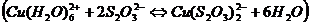 , оттитровывают цинк
, оттитровывают цинк ![]() и находят содержание меди по разности.
и находят содержание меди по разности.
Иодометрическое титрование[1]
Методика основана на взаимодействии меди с избытком иодида калия и последующем титровании выделившегося иода тиосульфатом
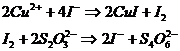
Определение возможно как в кислой, так и в нейтральной среде, в нейтральном растворе титрование идет медленнее и часто к концу его вновь появляется окрашивание; результаты занижены по сравнению с титрованием в слабокислых растворах. Присутствие небольших количеств соляной кислоты не мешает, с большим количеством возможно образование комплексов; мешают также железо(III), мышьяк(III) и сурьма(III), благородные металлы; окислители, способные окислять I-(ванадий(V), ![]()
![]()
![]()
![]() ,
, ![]() ); значительное количество хлорид-, бромид-ионов и комплексонатов, образующих комплексы с Cu(II). В качестве индикатора используют свежеприготовленный раствор крахмала. Присутствующий в сплаве цинк определению не мешает.
); значительное количество хлорид-, бромид-ионов и комплексонатов, образующих комплексы с Cu(II). В качестве индикатора используют свежеприготовленный раствор крахмала. Присутствующий в сплаве цинк определению не мешает.
3.1.2.Гравиметрическое определение
Осаждение салицилальдоксимом[1]
Осаждение проходит по уравнению
![]()
Полнота осаждения достигается при рН 2,6-3,3. При данном рН осаждение меди достаточно селективно: вместе с медью осаждаются только Fe(III) (которое можно предверительно осадить аммиаком или замаскировать винной кислотой) и Ni(II) (отделение в виде диметилглиоксимата). Остальные металлы, в том числе Zn, не мешают. После фильтрования и промывания осадок сушат при температуре 105-110°С. Гравиметрической формой служит Cu(C7H6O2N)2, совпадающая с осаждаемой формой.
Осаждение солью Рейнеке[2]
Осаждение проходит по уравнению
![]() ,
,
где Cu(II) предварительно восстанавливают до Cu(I) обычно хлоридом олова(II)
![]() .
.
Cu[Cr(NH3)2(CNS)4] нерастворим в разбавленных минеральных кислотах (при их концентрации не выше 3 М). Определению меди мешает присутствие Hg(I), Tl(I), Ag(I), образующих в этих условиях осадки аналогичного состава. После фильтрования и промывания осадок сушат при температуре 110°С. Гравиметрической формой служит Cu[Cr(NH3)2(CNS)4], совпадающая с осаждаемой формой.
3.3.Обсуждение выбора методики определения меди
Достоинства, недостатки и основные характеристики титриметрических и гравиметрических методик определения меди приведены в таблице 2.
Таблица 2(характеристики методик определения меди)
Методики Характеристики | Комплексонометрическое титрование с ЭДТА | Йодометрическое титрование | Осаждение салицилальдоксимом | Осаждение солью Рейнеке |
Растворение образца | Сильные кислоты в присутствии HNO3 | Сильные кислоты в присутствии HNO3, большие количества HNO3 необходимо затем удалить | Сильные кислоты в присутствии HNO3 | Сильные кислоты в присутствии HNO3, большие количества HNO3 необходимо затем удалить |
Установление необходимого значения рН | рН 5, ацетатный буфер | рН>2 | рН 2.6-3.3 | рН 1 |
Способ титрования | Прямой | Заместительный | - | - |
Структура осадка | - | - | Скрытокристаллический, подобный аморфному | Скрытокристаллический, подобный аморфному |
Индикатор | ПАН, ПАР | Раствор крахмала | - | - |
Мешающие ионы образца | Zn титруется совместно, потом отдельно, Cu находим по разности | - | - | - |
Гравиметрическая форма | - | - | Cu(C7H6O2N)2 | Cu[Cr(NH3)2(CNS)4] |
Перевод в конкретную степень окисления | - | - | - | Перевод Cu(II) в Cu(I) (K2SnCl4) |
Стехиометричность реакции | + | + | + | + |
Полнота протекания реакций [4] |
при рН 5 |
| ||
Гравиметрический фактор в пересчете на Cu | - | - | 0,1916 | 0,1636 |
Удобство фиксирования к. т.т. | Переход может быть нечетким из-за окраски CuY2- | Переход четкий | - | - |
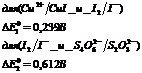
3.4.Обоснование выбора методики
При выборе методики определения меди мы сразу исключили гравиметрические методики, несмотря на то, что они селективны (присутствующий в образце цинк не мешает определению) и достаточно чувствительны (малое значение гравиметрического фактора). Однако реагенты, требуемые для осаждения малодоступны, методики длительны и трудоемки, а требуемые значения рН трудно установить (для области рН 1-3,3 нет буферных растворов). Кроме того, осаждение солью Рейнеке требует проведения дополнительной реакции восстановления Cu(II) в Cu(I). Высокая же чувствительность в данном случае не требуется, так как медь составляет основу сплава.
Что касается титриметрических методик, то их можно использовать для определения меди в данном сплаве. Они просты в выполнении, точны, а иодометрическая методика, кроме того, ещё и селективна(Zn определению не мешает).
Для определения меди были выбраны как иодометрическая, так и комплексонометрическая методики. При помощи последней определили и цинк.
При выполнении определения следует иметь в виду следующее:
медные сплавы быстро растворяются только в присутствии HNO3, поэтому азотную кислоту необходимо использовать при растворении, однако лучше добавлять её как можно меньше, а основные количества следует удалить, так как она может впоследствии окислять4.Определение меди в образце
4.1.Схема количественного анализа образца
![]()

![]()
4.2.Расчет навески
Титранты, применяемые в обоих титрованиях, имеют приблизительно одну и ту же концентрацию (0,05 М). Поэтому оба определения можно проводить из раствора одной навески. Мы исходили из того, что концентрация меди в титруемом растворе (с) должна составлять ~0,05 М; в этом случае на титрование аликвоты 10,00 мл пойдет примерно равное количество (~10 мл) титранта. Содержание меди в образце (ω) составляет ~60% масс., объем раствора для титрования (V) выбрали равным 200,0 мл, отсюда масса навески составляет
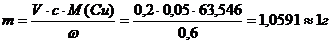
Взятие навески
масса, г | |
стаканчик со сплавом | 11,2601 |
стаканчик без сплава | 10,1991 |
m(навески)=1,0615г
4.3.Растворение пробы
Реагенты:
![]() 2М раствор
2М раствор
![]() (1:1) раствор
(1:1) раствор
![]() конц.
конц.
![]() 6М раствор
6М раствор
![]() конц. раствор
конц. раствор
![]() 1М раствор
1М раствор
Методика:
В стакан на 200 мл внесли навеску сплава, 30 мл H2SO4 2M, 20 мл HCl (1:1) и поставили на песчаную баню. Так как растворение шло крайне медленно, добавили 5 капель конц. HNO3. После испарения значительного количества воды и добавления HNO3 скорость реакции значительно увеличилась. По мере выкипания раствора добавляли HCl (1:1) и еще несколько капель HNO3 6М (для ускорения реакции), а также H2O для предотвращения кристаллизации образовавшихся солей. По окончании растворения образца в полученном растворе содержалось лишь незначительное количество нитрат-ионов, так как основное количество удалилось в виде NO2. Для понижения кислотности раствора добавили аммиак до начала выпадения осадка Cu(OH)2, затем растворили осадок, добавив несколько капель ![]() 1М. В итоге получили слабокислый раствор сплава, который затем разбавили дистиллированной водой до 200,0 мл.
1М. В итоге получили слабокислый раствор сплава, который затем разбавили дистиллированной водой до 200,0 мл.
![]()
![]()
4.4.Методики определения
4.4.1.Иодометрическое определение
Реагенты:
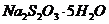 стандартный раствор (~0.05 М)
стандартный раствор (~0.05 М)
![]() 5% раствор
5% раствор
![]() 1 М раствор
1 М раствор
крахмал 1-% свежеприготовленный раствор
Методика:
- заполнить бюретку раствором ![]() и закрыть трубкой с натронной известью
и закрыть трубкой с натронной известью
- в коническую колбу для титрования на 100 мл внести пипеткой 10,00 мл анализируемого раствора, добавить 2мл раствора ![]() , 30 мл раствора
, 30 мл раствора ![]() и титровать до бледно-желтой окраски суспензии
и титровать до бледно-желтой окраски суспензии
- добавить к раствору 1-2капли раствора крахмала и титровать до белой окраски суспензии
![]()
4.4.2.Комплексонометрическое определение
Реагенты:
ЭДТА стандартный раствор (~0,05 М)
ацетатный буфер с рН 4,8-5,2
10% раствор
Металлоиндикатор ПАН 0,1% водный раствор
Методика:
- Определение суммы Cu и Zn
в коническую колбу на 200 мл внести 10,00 мл анализируемого раствора, 10мл ![]() , 10мл буферного раствора, нагреть на песчаной бане до ~70°С, затем добавить 10капель ПАН и титровать раствором ЭДТА до начала перехода окраски из фиолетовой к зеленую
, 10мл буферного раствора, нагреть на песчаной бане до ~70°С, затем добавить 10капель ПАН и титровать раствором ЭДТА до начала перехода окраски из фиолетовой к зеленую
- Определение Zn
В коническую колбу на 200мл внести 10мл анализируемого раствора, добавить раствор ![]() до обесцвечивания, 10мл буферного раствора, 10капель ПАН и титровать до изменения окраски из красной в оранжево-желтую
до обесцвечивания, 10мл буферного раствора, 10капель ПАН и титровать до изменения окраски из красной в оранжево-желтую
- Определение Cu
V(ЭДТА), пошедший на титрование Cu найти по разности V(ЭДТА) в отсутствии и в присутствии тиосульфата
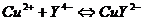
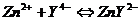
4.4.3Приготовление и подготовка реактивов
Приготовление и стандартизация раствора тиосульфата
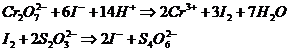
Реагенты:
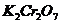 первичный стандартный раствор
первичный стандартный раствор
![]() 1М
1М
![]() 5%-й раствор
5%-й раствор
крахмал 1%-й свежеприготовленный раствор
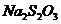 ~0,05М
~0,05М
Методика:
- в бюретку налить раствор ![]() и закрыть трубкой с натронной известью
и закрыть трубкой с натронной известью
- в коническую колбу на 200-250мл внести 10мл ![]() , 10мл
, 10мл ![]() , 10мл
, 10мл ![]() (отбор пипеткой) и оставить на 3-5мин в темном месте, прикрыв часовым стеклом
(отбор пипеткой) и оставить на 3-5мин в темном месте, прикрыв часовым стеклом
- затем добавить в раствор 100мл ![]() и титровать до бледно-желтой окраски суспензии
и титровать до бледно-желтой окраски суспензии
- прибавить несколько капель крахмала и титровать до белой окраски суспензии
Приготовление первичного стандартного раствора
Для приготовления 200,0 мл (V) 0,0500 М (с) 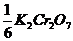 необходимо взять навеску
необходимо взять навеску
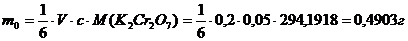
Взятие навески:
масса, г | |
стаканчик с веществом | 6,7274 |
стаканчик без вещества | 6,221 |
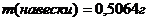
Поместить навеску в мерную колбу на 200,0 мл и растворить в минимальном количестве дистиллированной воды, затем довести раствор до метки, закрыть пробкой и тщательно перемешать.
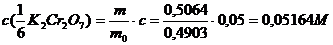
4.5.Результаты определения
Иодометрическое определение
V(Na2S2O3), мл
11.30
11,31
11,30
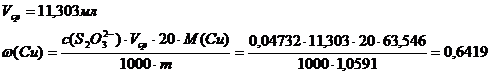
Комплексонометрическое определение
1.определение суммы Cu и Zn
Объем ЭДТА V1 (мл)
16,39
16,30
16,34
Vср=16,34 мл
2.определение Zn
Объем ЭДТА V2 (мл)
5,96
5,90
5,88
Vср=5,91 мл
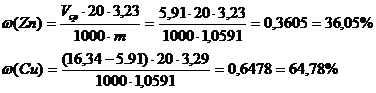
4.6.Описание особенностей или сложностей анализа
1. Поскольку значительные количества HNO3 мешают иодометрическому определению меди, мы первоначально приняли решение растворить образец вовсе без HNO3, в смеси (H2SO4 2 M 30 мл + HCl (1:1) 20 мл). Однако в этом случае растворение пошло крайне медленно. Поэтому к раствору добавили 5 капель конц. HNO3 и упарили для удаления основной массы воды. При этом скорость растворения резко увеличилась. В дальнейшем можно рекомендовать для растворения медных сплавов использовать небольшое (~10 мл) количество достаточно концентрированной (1:4 или 1:2) H2SO4 с добавками (по нескольку капель) конц. HCl и HNO3.
2. Раствор образца достаточно кислый, это может помешать как иодометрическому так и комплексонометрическому определению. Поэтому после растворения (до разбавления раствора до метки) мы понизили его кислотность, как описано выше. Кроме того, для комплексонометрического титрования взяли удвоенное по сравнению с рекомендуемым количество буферного раствора.
3. В ходе комплексонометрического определения при титровании Zn нагревание проводить нельзя, так как в растворе присутствует тиосульфат, но реакция комплескообразования идет относительно медленно, поэтому перед конечной точкой титрования необходимо немного подождать – изменения цвета индикатора может занять некоторое время, иначе результаты могут быть завышены.
4. Пред титрованием Zn медь маскируют тиосульфатом по реакции ![]() , но предложенного в методике количества тиосульфата (3-5 мл) явно недостаточно, так как при добавлении 5 мл цвет раствора не меняется (
, но предложенного в методике количества тиосульфата (3-5 мл) явно недостаточно, так как при добавлении 5 мл цвет раствора не меняется (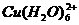 - голубой,
- голубой, 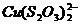 - бесцветный), поэтому было решено добавлять тиосульфат до обесцвечивания раствора. Объем раствора тиосульфата, пошедшего на маскирование меди, составил 50 мл.
- бесцветный), поэтому было решено добавлять тиосульфат до обесцвечивания раствора. Объем раствора тиосульфата, пошедшего на маскирование меди, составил 50 мл.
5. При титровании суммы Cu и Zn, замечено, что переход окраски из фиолетовой в зеленую нечеток, так как комплексонат CuY2- окрашен в синий цвет. Поэтому было решено добавить существенно большее количество индикатора (10 капель вместо 3 рекомендуемых методикой).
4.7.Выводы
Содержание Cu в образце составляет 64,19% по результатам иодометрического титрования и 64,78% по результатам комплексонометрического титрования. Содержание цинка по результатам комплексонометрического титрования составляет 36,05%.
5.Список литературы
1., . Аналитическая химия элементов. Медь. Москва: Наука, 1990
2.. Анализ руд цветных металлов. Москва: Металлургиздат, 1953
3.. Практическое руководство по гравиметрии и титриметрии. Москва: НЬЮДИАМЕД, 1996.
4.. Справочник по аналитической химии. Москва: Химия, 1971
