

Партнерка на США и Канаду по недвижимости, выплаты в крипто
- 30% recurring commission
- Выплаты в USDT
- Вывод каждую неделю
- Комиссия до 5 лет за каждого referral
Лекции для специальности « Фармация» 1 курс. Часть1
дисциплина: физическая и коллоидная химия
кафедра физической и коллоидной химии
лектор:доцент, к.х.н.
Физическая химия - многоотраслевая наука, изучающая взаимосвязь различных физических и химических свойств материи, включая исследование механизмов и скоростей химических реакций, зависимость их от внешних факторов, а также устанавливающая связь между химическими свойствами, составом и строением вещества.
Х и м и ч е с к а я т е р м о д и н а м и к а.
Основные понятия и определения. Первый закон термодинамики.
Термодинамика (ТД) – общая теория состояния макроскопических тел, которая из совокупности закономерностей, выведенных логическим путем, позволяет предвидеть изменение состояния любого макроскопического тела в результате его взаимодействия с окружающими телами.
Термодинамика построена по дедуктивному принципу: на основе двух законов, называемых началами термодинамики, которые обобщают опытные данные, выводятся следствия для различных частных случаев, что позволяет определить принципиальную возможность конкретного процесса и избежать постановки эксперимента, обреченного на неудачу. В отношении химических и биохимических процессов термодинамика позволяет решить вопрос о том, возможна ли данная реакция и каков может быть максимальный выход продуктов. Однако, термодинамика ничего не говорит о скорости процесса, и в этом главное ее ограничение.
Объектом изучения термодинамики являются макроскопические тела, состоящие из большого числа атомов или молекул. Тело, часть тела или группа тел, выделенные из пространства реально существующими или воображаемыми поверхностями, называются термодинамической системой. Остальная часть пространства образует внешнюю ( окружающую) среду. Пример ТД системы: газ в баллоне, кофе в чашке, кристалл, ваш рабочий стол, человеческий организм и т.д. или выделенная мысленно часть подобного рода объектов.
Классификация термодинамических систем.
Изолированные системы – системы, которые не обмениваются с окружающей средой ни энергией, ни веществом. Изолированные системы практически отсутствуют в природе, однако существует практическая целесообразность их рассмотрения, так как процессы в этих системах можно представить как идеальные, чтобы затем сравнить их с реальными. Пример: кабина космического корабля, подводной лодки.
Закрытые системы – системы, которые обмениваются с окружающей средой энергией, но не веществом. Пример: консервная банка, воздушный шарик…
Адиабатические системы – системы, которые обмениваются с окружающей средой только веществом. Пример: сосуд Дюара, термостат.
Открытые системы – системы, которые обмениваются с окружающей средой и энергией и веществом. Пример: человеческий организм, стакан с чаем, такое глобальное геополитическое образование как страна.
Макроскопические признаки, характеризующие систему, называются
термодинамическими параметрами. Наибольшее значение имеют параметры, поддающиеся непосредственному определению т.е. температура, давление, объем, плотность, концентрация и др.
Гомогенные системы - системы, внутри которой ТД параметры остаются постоянными или непрерывно меняются от точки к точке. Система называется гетерогенной, если она состоит из нескольких макроскопических частей, отделенных одна от другой видимыми поверхностями раздела. На этих поверхностях некоторые параметры меняются скачкообразно. Совокупность всех гомогенных частей системы, однородных в физическом и химическом отношениях, называется фазой. Агрегатные состояния (газ, жидкость, различные модификации кристаллов) представляют собой примеры разных фаз. Фаза может состоять из одного, а чаще из нескольких компонентов ( если фаза, например, раствор). Компонент – это химически индивидуальное вещество, которое может быть выделено из системы и существовать самостоятельно. Лекарства, продукты питания – сложные гетерогенные системы.
Состояния системы.
Если термодинамические параметры, характеризующие систему, изменяются с течением времени, то такое состояние системы называется неравновесным. Как показывают экспериментальные наблюдения, если внешние условия не меняются, то в системе постепенно прекращаются макроскопические изменения, и каждый параметр, характеризующий какое-либо макроскопическое свойство системы, приобретает постоянное во времени значение. Такое состояние системы называется равновесным.
Как показывает опыт, между различными химическими и физическими свойствами равновесной системы существуют определенные зависимости, а следовательно, термодинамические параметры, которые характеризуют эти свойства, функционально связаны между собой. Например, в закрытой гомогенной системе, состоящей из одного компонента, температура (Т), объем (V) и давление ( Р ) связаны функциональной зависимостью:
ƒ ( Р, Т, V ) = 0 (1.1)
Уравнение, связывающее термодинамические параметры системы в равновесном состоянии, называются уравнением состояния. Параметры, которые выбраны для однозначной характеристики системы , называются независимыми параметрами. В термодинамике уравнения состояния считаются известными из опыта.
Пример: Состояние сильно разреженного газа, состоящего из индивидуального вещества, расстояние между молекулами которого намного превышают их диаметр, что позволяет пренебречь энергией межмолекулярного взаимодействия по сравнению с энергией поступательного движения молекул, хорошо описывается уравнением:
P V = n R T (1.1a),
где R – универсальная газовая постоянная, равная 8.3143 Дж / К моль, n -число молей, заключенных в объеме V (м3) при давлении P ( Па ) и температуре T (К).
Газ, точно подчиняющийся уравнению (1.1а), наз. идеальным газом. Независимо от химической природы вещества при достаточно низких давлениях все газы подчиняются уравнению (1.1а), т.е это уравнение является предельным законом при описании реальных газов.
Классификация термодинамических параметров
Внешние параметры – величины, определяемые положением не входящих в данную систему тел. Например, объем системы определяется расположением внешних тел, напряженность внешнего силового поля зависит от положения источников поля, не входящих в данную систему.
Внутренние параметры- величины, определяемые движением и расположением в пространстве частиц, входящих в данную систему. Давление, плотность, поляризация зависят от взаимного расположения частиц системы, скорости их движения, сил взаимодействия между ними. Однако считать тот или иной параметр внешним или внутренним зависит от того, как мы проводим границу между системой и окружающей средой.
Другая классификация ТД параметров связана с их зависимостью от размеров системы:
Интенсивные параметры не зависят от размера системы. Это - температура, давление и т.д. Переменные, значения которых изменяются пропорционально размерам и массе системы при ее разбиении на части без нарушения равновесного состояния, называются экстенсивными параметрами. К ним относятся объем, длина, масса и т.д. Экстенсивные параметры обладают свойством аддитивности. Интенсивные параметры могут быть измерены лишь через некоторую экстенсивную величину.
Первый закон термодинамики.
Первый закон термодинамики (первое начало термодинамики) является применением закона сохранения и превращения энергии к термодинамическим системам. В основе этого закона лежит многовековой опыт человечества и бесчисленные эксперименты, доказывающие сохранение энергии при всех ее превращениях. Первый закон термодинамики имеет несколько формулировок, общий смысл которых один и тот же. Приведем три формулировки:
· Энергия не создается и не уничтожается.
· Внутренняя энергия системы является однозначной функцией состояния.
· Теплота, поглощенная системой и совершенная над системой работа расходуются на увеличение внутренней энергии этой системы.
При бесконечно малых изменениях, происходящих в закрытой системе, первое начало термодинамики можно представить в виде:
dU = dQ + dW ( 1.2 )
В интегральном виде:
DU = Q + W ( 1.3 )
Для открытой системы обмен с окружающей средой веществом первый закон термодинамики запишется следующим образом:
dU = dQ + dW+ dEm ,где (1.4a) последнее слагаемое учитывает обмен веществом системы с окружающей средой. В случае изолированной системы первый закон термодинамики имеет вид:
dU=0 (1.4б)
На термодинамическую систему могут действовать различные внешние силы, которые совершают работу. В зависимости от того, какие силы действуют на систему, работа может быть механической, электрической, магнитной, гравитационной и т.д. Механическая работа ( работа расширения или сжатия ) выражается формулой:
dW = - pdV, где р- давление (1.5)
Первый закон термодинамики можно записать с учетом разделения работы на механическую работу и прочие виды работ:
dU = dQ + dW¢- pdV (1.6)
Если прочие виды работ отсутствуют, т.е. dW¢ = 0, то уравнение 2.5 может быть записано следующим образом:
dU = dQ ¢- pdV (1.7)
Применение первого закона термодинамики к различным процессам
Теплоемкость. Термохимия. Закон Гесса.
Рассмотрим гомогенную однокомпонентную закрытую систему
1. В изотермическом процессе ( Т = Const.) внутренняя энергия будет оставаться постоянной:
dQ + dW=0 (1.8)
Следовательно, если система получает тепло, то она совершает работу. Если над системой совершают работу, то система отдает тепло окружающей среде.
2. В изохорическом процессе, т.е. при V = Const.,
dV = 0
В случае закрытой системы, где учитывается только работа расширения
( сжатия) из ( 1.8 ): dU = dQV ( 1.9 )
Теплота в этих условиях не зависит от пути процесса и становится функцией состояния.
Так как система гомогенная и однокомпонентная, то из уравнения
dU = ( ¶U/¶V)T dV + (¶U/¶T)VdT, при V=Const., получим
dQV = (¶U/¶T)VdT, (1.10)
Обозначим: CV = (¶U/¶T)V = dQV/dT, где СV – теплоемкость при постоянном объеме, т.е. количество теплоты, необходимое для повышения температуры системы на один градус при постоянном объеме. Отсюда изменение внутренней энергии можно рассчитать по формуле:
dU = CVdT (1.11)
3. В изобарическом процессе, т.е. при Р = Const.,
dP = 0.
Перепишем (2.4): dQ = dU + pdV, и при Р = Const.:
dQ = dU + d (pV) = d ( U+ pV)
Введем обозначение: Н º U + pV, (1.12)
которое определяет новое свойство системы - энтальпию, имеющее размерность энергии и являющееся функцией состояния. Теперь
dH = dQP (1.13)
Теплота в этих условиях не зависит от пути процесса и становится функцией состояния.
Для гомогенной однокомпонентной системы энтальпия будет однозначно определена, если заданы два параметра состояния, например Т и Р:
Н= Н( Т, Р) (1.14)
Тогда полный дифференциал энтальпии:
dН = ( ¶Н/¶Р)T dР + (¶U/¶T)РdT, при Р = Const.
dQР = (¶Н/¶T)РdT (1.15)
Обозначим CР = (¶Н/¶T)Р = dQР/dT, где СР – теплоемкость при постоянном давлении, т.е. количество теплоты, необходимое для повышения температуры системы на один градус при постоянном давлении. Отсюда изменение энтальпии можно рассчитать по формуле:
dН = CРdT (1.16)
Для идеального газа в случае мольных величин можно показать, что
CР = CV + R (1.17)
где R – универсальная газовая постоянная, имеющая смысл работы, совершаемой одним молем идеального газа в изобарическом процессе при нагревании на один градус.
Таким образом в изохорическом и изобарическом процессах все количество теплоты, полученное системой, идет на изменение ее внутренней энергии или энтальпии соответственно, а последние характеризуют тепловой эффект химических реакций. Наука о тепловых эффектах, сопровождающих химические и физические изменения веществ наз. термохимией.
Формулы (1.9) и (1.13) являются математическим выражением закона Гесса ( закон постоянства сумм теплот реакции): если из данных исходных веществ можно различными путями получить заданные конечные продукты, то независимо от пути получения , вида промежуточных реакций, суммарный тепловой эффект для всех путей будет одним и тем же, если:
- единственной работой, совершаемой системой, является работа сил внешнего давления ( работа расширения или сжатия);
- давление или объем в течение всего процесса остаются неизменными,
- полученные продукты имеют одну и ту же температуру, что и исходные вещества;
- процесс протекает термодинамически неравновесно (необратимо).
Тепловой эффект, сопровождающий любую химическую реакцию, может бать выражен двумя способами: термохимическим и термодинамическим. Как правило, в дальнейшем мы будем пользоваться последним:
Например, для эндотермической реакции запись
3C + 3H2 = C3 H 2 – 20.5 кДж/моль является термохимической, а
3C + 3H2 = C3 H 2, DН = 20.5 кДж/моль – термодинамической.
Большое значение закона Гесса заключается в том, что пользуясь им, можно вычислить неизвестную теплоту реакции путем комбинирования стехиометрических уравнений реакций и их тепловых эффектов. Для расчетов также широко используются два следствия, вытекающие из закона Гесса.
1. Тепловой эффект химической реакции равен сумме теплот образования продуктов реакции за вычетом сумм теплот образования исходных веществ:
DН = å( niDНобраз. прод. ) - å( ni DНобраз. исх.в-в ) (1.18а)
2. Тепловой эффект химической реакции равен сумме теплот сгорания исходных веществ реакции за вычетом сумм теплот сгорания продуктов реакции.
DН = å( ni DНсгор.исх.в-в ) - å( ni DНсгор.прод.) , где (1.18б)
ni – стехиометрические коэффициенты соответствующих веществ в уравнении химической реакции.
Стандартные тепловые эффекты.
Тепловые эффекты химических реакций существенно зависят от температуры и в меньшей степени от давления, поэтому сопоставимы тепловые эффекты, отнесенные к одинаковым условиям. В целях удобства условились относить тепловые эффекты к определенным стандартным условиям, т.е. считать, что реакция осуществляется между веществами, находящимися в стандартных состояниях. Стандартные состояния твердых и жидких веществ – это устойчивое состояние чистого вещества при данной температуре под давлением 1 атм. Тепловые эффекты процессов, определенные при участии в этих процессах веществ, находящихся в стандартных состояниях, носят название стандартных тепловых эффектов - DН° или DU° ( дельта « аш» стандартное или дельта « у » стандартное). Стандартные тепловые эффекты могут относится к любой температуре, однако большинство современных данных, приведенных в справочниках, отнесены к температуре 298К и обозначены DН°298.
Стандартная теплота образования – теплота образования одного моля соединения из простых веществ или элементов, которые находятся в стандартных состояниях.
Стандартная теплота сгорания неорганических веществ – тепловой эффект реакции окисления одного моля данного соединения кислородом с образованием высших оксидов соответствующих элементов.
Стандартная теплота сгорания органических веществ – тепловой эффект реакции полного сгорания в кислороде данного соединения до жидкой воды, диоксида углерода и высших оксидов гетероатомных элементов.
Зависимость теплового эффекта химической реакции от температуры.
( Формула Кирхгофа)
Формула Кирхгофа позволяет рассчитать тепловой эффект реакции при любой температуре, если эта величина известна для одной температуры и известны также мольные теплоемкости компонентов, участвующих в реакции.
В общем случае, если реакция протекает при постоянном давлении, то
DН2 Т2
òd (DН ) = DН2 - DН1 = ò (å ni D Cp ) dT ( 1.19)
DН1 Т1
Если теплоемкость веществ – участников реакции не зависит от температуры, то формула (1.19) имеет вид:
DН2 = DН1 + DСр ( Т2 - Т1 ) ( 1.19а)
Самопроизвольные и несамопроизвольные процессы.
Второй закон термодинамики.
Второй закон термодинамики, который постулируется на основании многовекового человеческого опыта, дает возможность предсказать направление процесса. Именно этот закон позволяет разделить все процессы, которые возможны с точки зрения первого закона термодинамики, на две различные группы: самопроизвольные и несамопроизвольные.
Самопроизвольные процессы – это неравновесные процессы, которые протекают без воздействия внешней силы в направлении достижения равновесия. Для проведения самопроизвольных процессов не только не затрачивается работа, но и при соответствующих условиях эта система сама может произвести работу в количестве, пропорциональном происходящему изменению. Пример: переход тепла от более нагретого тела к менее нагретому, смешение газов, расширение газов а вакуум и т.д.
Несамопроизвольные процессы – процессы, удаляющие систему от состояния равновесия, которые не могут происходить без внешнего давления, т.е. для проведения таких процессов необходимо затратить работу в количестве, происходящим изменениям.
Равновесные процессы – процессы, при которых система, бесконечно медленно изменяясь, проходит непрерывный ряд одних и тех же равновесных состояний в прямом и обратном направлениях. Равновесные процессы – это обратимые процессы. Их можно рассматривать в качестве промежуточных между самопроизвольными и несамопроизвольными.
Формулировок второго закона термодинамики около сорока. Остановимся на некоторых из них:
· Единственным результатом любой совокупности процессов не может быть переход теплоты от менее нагретого тела к телу более нагретому.
· Существует некоторое экстенсивное свойство системы S, называемое энтропией, изменение которого следующим образом связано с поглощаемой теплотой и температурой системы:
- в неравновесном процессе dS > dQ/T; (2.1)
- в равновесном процессе dS = dQ/T; (1.2)
Отношение теплоты, поглощенной системой, к температуре наз. приведенной теплотой.
Термин « энтропия » был впервые введен Клаузиусом в 1865 г. Буквальный перевод с греческого этого слова означает « превращение в », т.е. имелась
ввиду тенденция превращения энергии в менее ценные формы.
В случае отсутствия теплообмена с окружающей средой:
dS ³ 0 ; (2.3)
или в интегральной форме:
S2 - S1 ³ 0 - энтропия адиабатной системы постоянна в равновесных процессах и возрастает в неравновесных. Адиабатические процессы называются поэтому также изоэнтропными.
Следовательно, исследуя изменение энтропии в изолированных или адиабатных системах, можно предсказать направление процесса. Если в исследуемом процессе энтропия системы будет увеличиваться. То такой процесс в системе может протекать самопроизвольно. Убыль энтропии означает невозможность данного процесса. Постоянство энтропии характеризует состояние равновесия. Так как в самопроизвольном процессе энтропия увеличивается, то при наступлении равновесия энтропия будет максимальна.
Таким образом, деление процессов на самопроизвольные и несамопроизвольные нашло свое количественное выражение: DS>0 – критерий осуществления самопроизвольного процесса в адиабатическом процессе и в изолированной системе.
Вычисления изменений энтропии в различных процессах.
Энтропия является функцией состояния системы, и поэтому изменение энтропии будет одинаково при равновесном и неравновесном переходе системы из одного состояния в другое. Так как не существует приборов, позволяющих измерить изменение энтропии, эту величину можно только рассчитать для равновесного процесса по уравнению (2.2) .
1. Изохорический процесс:
dS = dQ / T= dU / T = CvdT / T (2.4)
2
DS = ò Cv dT / T ( 2.4а)
1
2. Изобарический процесс:
dS = dQ / T= dН / T = CрdT / T (2.5)
2
DS = ò Cp dT / T (2.5а)
1
В обоих случаях необходимо знать зависимость теплоемкостей от температуры.
3. Изотермический процесс ( для гомогенной системы, состоящей из идеального газа):
DS = dQ / T = pdV/T = nRTdV/VT (2.6)
2
DS = nR ò dV / V = nR ln V2/ V1 (2.6a)
1
4.Фазовый переход. При фазовых переходах температура остается постоянной.
dS = dQ / T
2 2
DS = ò dQ / T = 1/ T = ò dQ = Qt / T , где
1 1
Qt - это теплота фазового перехода. Но так как фазовые превращения, такие как плавление, испарение, возгонка, превращение кристаллических модификаций и др., проходят и при постоянном давлении, то
Qt = DН ф. перехода ( 2.7)
DSф.перехода = DН ф. перехода/ Т ( 2.7а)
Постулат Планка.
На основании изучения большого числа экспериментальных данных Макс Планк постулировал: Энтропия индивидуального вещества, кристаллы которого идеально построены, при абсолютном нуле равна нулю.
lim S = 0 (2.8)
T® 0
При идеально построенном кристалле все узлы его решетки заняты правильно чередующимися и закономерно ориентированными молекулами или ионами. Такие кристаллы называются идеальными твердыми телами. Реальные кристаллы таковыми не являются. Нарушения закономерностей совершенно естественны при высоких температурах, но они в какой-то мере сохраняются при охлаждении и « замораживаются» при абсолютном нуле.
В соответствии с постулатом Планка энтропию индивидуального твердого вещества можно вычислить по формуле:
T
S = ò CpdT / T (2.9)
0
Зная зависимость теплоемкостей тел в жидк5ом и газообразном состояниях, а также теплоты фазовых переходов, можно рассчитать абсолютную энтропию тела при любой температуре по формуле:
Tпл. Т кип. Т
S = ò Cp (тв.)dT / T + DHпл./ Тпл. + ò Cp (жид.)dT / T + DHкип../ Ткип. + ò Cp (газ.)dT / T
0 Тпл. Ткип
Термодинамические потенциалы.
Метод термодинамических потенциалов предложил американский физик Джосайа Уиллард Гиббс более ста лет назад, и с тех пор в него даже не вносились коррективы - настолько совершенным он является.
Термодинамический потенциал – величина, убыль которой определяет производимую системой работу. Но в термодинамике работа зависит от пути процесса, поэтому в каждом случае надо оговаривать условия проведения процесса. Кроме того, только в равновесном процессе можно получить максимальную работу. Именно с такой работой связывают величину термодинамического потенциала. В неравновесном процессе работа, совершаемая системой, будет всегда меньше убыли соответствующего термодинамического потенциала.
Второй закон термодинамики в общем виде может быть записан так:
dS ³ dQ/T , где количество теплоты можно выразить
на основании первого закона термодинамики. Тогда получим:
TdS ³ dU - dW (2.10)
Уравнение (3.10) отвечает объединенному первому и второму законам термодинамики и называется фундаментальным уравнением Гиббса.
Так как знак равенства относится к равновесным процесса, а при этом системой совершается максимальная работа, то
TdS = dU - dWравн. ,
Если же процесс неравновесный, то
TdS > dU - dW
В соответствие с ( 2.5) работа делится на работу расширения или сжатия
(-pdV)и на прочие виды работы (полезную работу dW). Тогда для равновесного процесса:
TdS = dU - dWравн. + pdV, (2.10а)
а для неравновесного процесса :
TdS > dU - dW + pdV, (2.10б)
Сравнивая (3.10а) и (3.10б) получим:
| dW| < | dWравн|
Если процесс протекает при S = Const., V = Const.( изохорно - изоэнтропийный ) процесс, то из уравнения ( 3.10а) следует :
- dWравн.= -dU
-Wравн.= - DU (2.10в)
Убыль внутренней энергии в определенных условиях характеризует полезную работу, следовательно, внутренняя энергия является изохорно-изоэнтропийным термодинамическим потенциалом Если система не совершает максимальной работы, то процесс ее изменения неравновесный, а его самопроизвольность определяется убылью соответствующего термодинамического потенциала:
- при переменных S и V из ( 2.10б) : dU < TdS – pdV ( 2.11)
если S = Const., V = Const. Из (2.11) получим dU <0 (2.11a)
Неравенство ( 2.11а) является термодинамическим условием протекания самопроизвольного процесса при S,V = Const.
- при переменных S и P можно показать: dH < TdS + Vdр ( 2.12)
Энтропия является изобарно- изоэнтропийным потенциалом и в условиях S = Const., P = Const. убыль ее характеризует возможность протекания самопроизвольного процесса:
dH < 0 ( 2.12а )
Выражения (2.11) и ( 2.12 ) позволяют ввести еще два термодинамических потенциала. Пусть в ( 2.11) только V = Const. , тогда dU < TdS , соответственно из (2.12 ) при постоянстве Р имеем dH < TdS . Таким образом в условиях Т, Р = Const.: dH – TdS < dG
или dG < dH – TdS (2.13)
а при T,V = Const.: dU – TdS < dA или dA< dU – TdS (2.14), где
G и A – функции состояния системы, бесконечно малые значения которых характеризуют разницу между тепловым эффектом полностью неравновесного процесса ( dU и dH) и теплотой нагрева системы ( TdS ) от абсолютного нуля до температуры Т. Последний вид энергии называется связанной энергией , контролируется энтропией системы. Функции G и A определяют ту часть энергии, которую система может отдать и превратить в работу, т.е. свободную энергию. «G» называется свободной энергией Гиббса, а «A » - свободной энергией Гельмгольца. В соответствующих условиях эти функции состояния становятся термодинамическими потенциалами и характеризуют максимально производимую работу в равновесных процессах, а в неравновесных – возможность протекания самопроизвольного процесса. Можно показать, что
при T,P dG < VdP – SdT , a если Т,P = Const. ,то dG < 0 (2.15)
при T,V dU< -PdV – SdT , a если T,V = Const., то dU < 0 (2.16)
Для равновесных процессов в случае конечных изменений выражения ( 2.13) и ( 2.14) превращаются в равенства :
DG = DH – TDS (2.17)
DA = DU - TDS (2.18)
Минимальное значение любого термодинамического потенциала в соответствующих условиях отвечает равновесию системы:
Условия состояние максимальная самопроизвольный
процесса равновесия работа процесс
Т,P = Const. dG = 0 (DG = 0) -Wравн.= - DG dG < 0 (DG < 0)
T,V = Const. dA = 0 (DA = 0) -Wравн.= - DA dA< 0 (DA < 0)
S,V = Const. dU = 0 (DU = 0) -Wравн.= - DU dU < 0 (DU < 0)
S,P = Const. dH = 0 (DH = 0) -Wравн.= - DH dH < 0 (DH <0)
V,U = Const. dS = 0 (DS = 0) -Wравн.= - DS dS > 0 (DS > 0)
Э л е к т р о х и м и я.
Электролиты - вещества, которые в растворе или в расплаве распадаются на ионы – электрически заряженные частицы, способные к самостоятельному существованию в этих средах. Электролиты – проводники второго рода .
Биожидкости организма, внутри- и межклеточные жидкости содержат большие количества электролитов, играющих важную роль в функционирование живых клеток. Биохимические процессы в организме протекают при непосредственном участии электролитов: они ответственны за проведение нервных импульсов, концентрацией электролитов во многом определяется проницаемость биологических мембран. Анализ содержания электролитов в тканях и жидкостях организма позволяет проводить диагностику патологических состояний, определить константы биохимических процессов.
Процесс распада растворенного вещества на ионы называется электролитической диссоциацией. Диссоциация наблюдается в полярных растворителях и зависит от e - диэлектрической проницаемости среды (чем больше e, тем диссоциация лучше).
Чаще всего примером электролитической диссоциации могут служить водные растворы. Схематически распад молекулы на ионы можно представить следующим образом:
1) ![]() (NaCl) для ионного типа решетки
(NaCl) для ионного типа решетки
2) ![]() (HCl) для неионного типа решетки с ковалентной связью.
(HCl) для неионного типа решетки с ковалентной связью.
Электропроводность растворов электролитов.
Электропроводность – способность веществ проводить электрический ток. Это – абстрактное понятие. А вот обратная величина, то есть неспособность раствора проводить электрический ток – сопротивление, можно измерить:
![]() [ См ] или [1/Ом ],
[ См ] или [1/Ом ],
где W – электропроводность, а R – сопротивление.
Удельная электропроводность k ( каппа ) – характеризует электропроводность раствора электролита объемом 1 м3, заключенного между двумя электродами, которые расположены на расстоянии ( l ) 1 м и имеет площадь поперечного сечения (S) 1 м2.
![]() , ( 3.1 )
, ( 3.1 )
где r - удельное сопротивление: ![]() [Ом ∙ м],
[Ом ∙ м],
Удельная электропроводность зависит от концентрации раствора, температуры, природы вещества:
1) 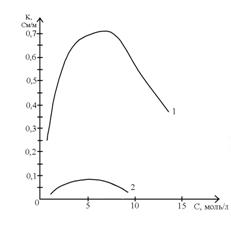 k = k (С)
k = k (С)
Рис.1 Зависимость удельной электропроводности
растворов от концентрации раствора
для сильного (кривая 1 ) и слабого ( кривая 2 ) электролитов.
2) Увеличение температуры на 1 0С, увеличивает электропроводность на 2-2.5 %.
Но в 1 м3 раствора разные электролиты содержат различное количество растворенного вещества, поэтому сложно сравнивать их удельные электропроводности. Для этого используется l – мольная электропроводность – электропроводность 1 моля раствора электролита, заключенного между двумя пластинами, расположенными на расстоянии 1 м.
![]() . ( 3.2.1 )
. ( 3.2.1 )
Если привести к 1 л объема, то:
![]() , ( 3.2.2 )
, ( 3.2.2 )
где n - разведение (разбавление), объем раствора, содержащий 1 моль вещества.
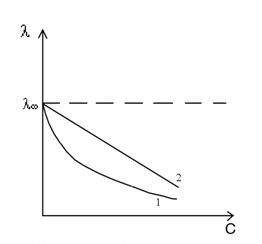 а б
а б
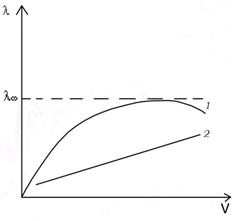
Рис. 2 Зависимость мольной электропроводности от концентрации ( а ) раствора и от разведения (б ) для слабого (кривая 1 ) и сильного (кривая 2) электролитов .
Основные положения теории Аррениуса (теория слабых электролитов).
1. Диссоциация молекул электролитов происходит уже в процессе растворения, когда диполи воды гидратируют молекулу растворенного вещества, что приводит к ее диссоциации за счет поляризации связи и превращения ее в ионную. Ионы существуют в растворе независимо от того, наложено на него или нет электрическое поле.
2. Растворы электролитов подчиняются законам разбавленных растворов. Растворенное вещество самопроизвольно распространяется в объеме растворителя подобно тому, как газ распространяется в пустоту (но характер их различен). Важно, что для разбавленных растворов электролитов закон действующих масс применим в том же виде, что и для идеальных газов.
3. Динамический характер диссоциации – в растворе непрерывно происходят многократные акты диссоциации молекул на ионы и соединения ионов в молекулы. Электролитическая диссоциация – равновесный обратимый процесс. Количественно характеризуется степенью электролитической диссоциации (a) и константой диссоциации (Кд), которые определяются законом действующих масс. Степень диссоциации зависит от природ растворителя и электролита. По величине степени диссоциации все электролиты делятся на слабые и сильные: если a < 0.03, то электролит считается слабым, если a > 0.3, то сильным. Степень диссоциации зависит от концентрации электролита. Если a = 1, то в растворе присутствуют только одни ионы.
a = a (С)
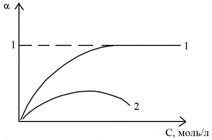
Рис. 3 Зависимость степени диссоциации от концентрации для
сильного ( кривая 1) и слабого ( кривая 2) электролитов.
Способность вещества диссоциировать в растворе учитывает и i – изотонический коэффициент Вант-Гоффа, характеризующий эффективное число частиц в растворе электролита, которое оказывает влияние на процессы в растворах электролитов.
![]() ,
,
который следующим образом связан со степенью диссоциации:
![]() ,
,
где n – число ионов на которых диссоциирует электролит. Для идеальных растворов i > 1.
Для электролитов коллегативные свойства учитывают наличие диссоциации:
Пример: 1) Осмотическое давление для электролитов : 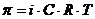
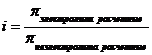 ,
,
2) закон Рауля для электролитов:
![]() .
.
4. В случае слабых электролитов, подвижности ионов не зависят от концентрации электролита, а l и l¥ ( мольная электропроводность при бесконечном разведении) различаются потому, что общее число ионов в растворе зависит от степени диссоциации:
![]() ,
,
![]() , (3.3 )
, (3.3 )
Пример: Рассмотрим диссоциацию типичного слабого электролита:
![]()
По закону действующих масс:
![]() ,
,
где ![]() ,
, 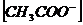 равновесная концентрация ионов
равновесная концентрация ионов ![]() и
и ![]() соответственно.
соответственно.
Пусть C0 – концентрация кислоты до диссоциации,
тогда
![]() ,
,
![]() .
.
![]() - Закон разбавления Оствальда. ( 3.4 )
- Закон разбавления Оствальда. ( 3.4 )
Для очень слабого электролита a << 1.Поэтому, когда a находится в знаменателе, ей можно пренебречь.
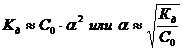
Сильные Электролиты фактически нацело диссоциируют на ионы, поэтому закону действующих масс и закону разбавления Оствальда не подчиняются. Следовательно, Кд зависит от концентрации при заданной температуре и не является постоянной. Применимость закона Оствальда является признаком слабого электролита.
Сильные электролиты. Теория Дебая-Хюккеля.
1. Ионы считаются лишенными размеров.
2. Теория учитывает лишь кулоновское взаимодействие между частицами.
3. Учитывают лишь электростатическое взаимодействие между центрами ионов и их ионной атмосферой.
Релаксационный эффект: Каждый ион окружен ионами противоположенного знака, которые образуют ионную атмосферу. Под действием электрического поля центральный ион выходит из ионной атмосферы, таким образом, разрушает ее и образует новую. возникает ее деформация, и возникает торможение иона за счет неравномерности ионной атмосферы. Время, необходимое для образования и разрушения ионной атмосферы называется временем релаксации.
Электрофоретический эффект: торможение ионной атмосферы под действием электрического поля.
Таким образом, сильные и слабые электролиты отличаются не только степенью диссоциации, но и всем механизмом диссоциации.
Подвижность ионов и числа переноса.
Электропроводность зависит от подвижности ионов. Молярную электропроводность раствора электролита можно выразить следующим образом:
![]() , где
, где
l+ и l- - мольная проводимость ионов или подвижность ионов, связанная с абсолютной скоростью движения ионов ![]() , которая зависит от размеров ионов и окружающей ионы сольватной оболочки, от валентности (для многовалентных ионов).
, которая зависит от размеров ионов и окружающей ионы сольватной оболочки, от валентности (для многовалентных ионов).
![]() ( 3.5 )
( 3.5 )
Подвижность имеет размерность мольной электропроводности 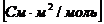 .
.
Пример:
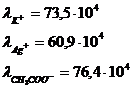
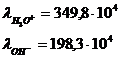
Закон Кольрауша (закон независимости движения ионов при бесконечном разбавлении).
![]() ( 3.6)
( 3.6)
При бесконечном разбавлении мольная электропроводимость равна сумме подвижностей ионов (ионных электропроводностей). Закон Кольрауша позволяет рассчитать предельную молярную электропроводность раствора электролита l¥ по известным значениям ионных подвижностей.
![]()
Пример:
![]()
![]()
![]()
![]()
Так как скорость ионов различна, следовательно, различная доля вещества, переносимая ими.
Число переноса – отношение абсолютной скорости движения катиона или аниона (u+ или u-) и суммарной скорости ионов:
![]() (3.7)
(3.7)
или
![]()
или
![]() ,
,
![]() (для слабого и для сильного электролита).
(для слабого и для сильного электролита).
Электродные потенциалы и электродвижущие силы.
Цепь проводников, включающая хотя бы один ионный проводник, называется гальванической. В разных звеньях гальванической цепи заряд переносится разными носителями – электронами или ионами.
Потенциометрия – совокупность физико-химических методов исследования, основанных на измерении ЭДС ( электродвижущей силы) в гальванических цепях. Это быстрый и во многих случаях незаменимый метод определения физиологически активных ионов (![]() ) в биологической жидкости (крови, спинномозговой жидкости) или тканях организма.
) в биологической жидкости (крови, спинномозговой жидкости) или тканях организма.
Процесс возникновения скачка потенциала на границе раздела двух фаз.
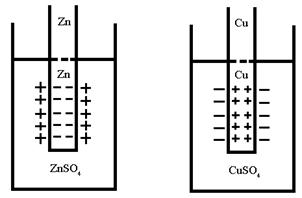
Структура металла – система плотно упакованных сфер одинакового радиуса – положительных ионов. Атомы металла располагаются в кристаллической решетке на таком расстоянии, что их валентные электроны фактически связаны не с отдельным ядром, а делокализованы (обобщены) по всем атомам. Суммарный положительный заряд равен числу электронов и в обычном состоянии металл электронейтрален.
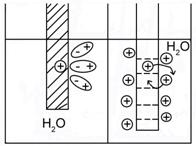
Растворение металла – переход катионов в раствор с поверхности электрода (пластинки металла) объясняется гидратацией катионов (Рис.4.). Катионы поверхности металла взаимодействуют с диполями воды. Возрастающая при этом теплота гидратации идет на разрыв связи ионов с кристаллической решеткой металла. Способность металла переходитьв раствор выражается тем сильнее, чем меньше потенциал ионизации и чем выше его ![]() .
.
Обратный процесс – осаждение катионов металла из раствора на поверхность электрода. Процессы перехода катионов металлов в раствор (окисление) и осаждение (восстановление) идут одновременно. Через определенный промежуток времени наступает динамическое равновесие на
границе раздела твердой и жидкой фаз.
Рис.4 Образование двойного электрического
слоя на границе металл-раствор.
Ионы из-за электростатического взаимодействия с противоположно заряженной поверхностью электрода ориентируются вблизи поверхности раздела фаз. Образуется двойной электрический слой (ДЭС) .
![]()
![]()
Рис. 5. Межфазный обмен электронами и электродные реакции.
Уравнение Нернста
Таким образом, на границе раздела двух фаз возникает разность потенциалов φ -гальвани-потенциал, величина которого зависит от природы контактирующих проводников. Величина φ определяется полезной работой самопроизвольного процесса, протекающего на границе раздела фаз: затрата химической работы компенсирована ростом скачка потенциалов Е (φ).
![]() ,
,
где n – число электронов электро-химического процесса, а φ – гальвани- потенциал.
![]() полуэлемент
полуэлемент
![]() для реакции:
для реакции:
По уравнению изотермы химической реакции:
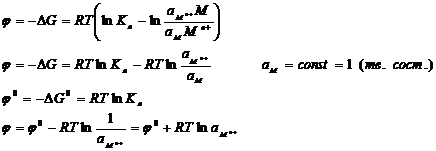
Выражение ( 3.8) – Уравнение Нернста.
Методов прямого измерения гальвани-потенциалов не существует. Для этого используют электродный потенциал. С этой целью составляют гальванический элемент из изучаемого электрода и электрода сравнения, потенциал которого известен, практически постоянен и хорошо воспроизводится. В качестве электродов сравнения чаще всего используются водородный, хлорсеребряный, каломельный электроды. Электродвижущая сила, возникающая в созданном гальваническом элементе (ЭДС) позволяет определить электродный потенциал.
Ряд напряженностей металлов – значения стандартных электродных потенциалов по отношению к стандартному потенциалу водородного электрода, расположенных в определенной последовательности.
![]()
 ,
,  ,
, 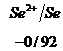 ,
, 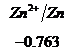 ,
, 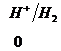 ,
, 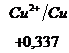 ,
, 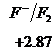
Гальванические элементы – химические источники электрического тока, позволяют получить работу за счет протекания химической реакции. Если из нескольких проводников (металлов растворов электролитов) составить цепь, то между концами такой правильно разомкнутой цепи возникает разность потенциалов Е ( ЭДС ), численно равная сумме всех скачков потенциалов на границах раздела фаз: ![]() . ЭДС – разность потенциалов на концах разомкнутой цепи, когда во внешней цепи сила тока равна нулю. В этом случае система находится в состоянии равновесия и
. ЭДС – разность потенциалов на концах разомкнутой цепи, когда во внешней цепи сила тока равна нулю. В этом случае система находится в состоянии равновесия и ![]() - максимально возможная работа по переносу единичного заряда вдоль замкнутой цепи. Единицы измерения ЭДС – вольты (В).
- максимально возможная работа по переносу единичного заряда вдоль замкнутой цепи. Единицы измерения ЭДС – вольты (В).
Условия работы гальванического элемента:
- пространственное разделение процессов;
- межфазный обмен электронами (то есть электродная реакция);
- протекание окислительно-восстановительной реакции по электрохимическому механизму.

Рис.6
![]() (3.9), где
(3.9), где
![]() - контактные разности потенциалов, которыми можно пренебречь.
- контактные разности потенциалов, которыми можно пренебречь.
Устройство и работа гальванического элемента Якоби – Даниэля
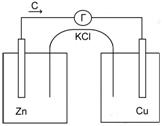
Рис.7 Устройство медно-цинкового гальванического элемента.
![]()
![]()
![]()
Гальванический элемент производит работу за счет окислительно-восстановительной реакции и поэтому его ЭДС, которая численно равна максимальной работе по перемещению заряда по замкнутой цепи, должна быть больше нуля. Из (3.9):
![]()
![]()
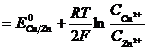
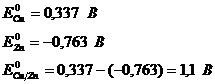
Классификация электродов
Электроды первого рода – металлы или неметаллы, погруженные в раствор, которые содержат их ионы с различной концентрацией.
![]()
Реакция на границе металл-раствор: ![]()
По уравнению Нернста: 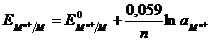 .
.
Электроды первого рода могут быть обратимыми как относительно катиона, так и относительно аниона: обратимость по катиону – электрод работает на основе обмена катионов между электродом и раствором (характерна для металлических электродов); обратимость по аниону – электрод работает на основе обмен анионов между электродом и раствором (характерна для неметаллических электродов и водородного). Иногда водородный электрод выделяют в газовые (инертный металл в контакте с раствором, содержащий ионы газообразного вещества, пропускаемого через раствор): водородный, хлорный, кислородный.
Рис. 8. Водородный электрод.
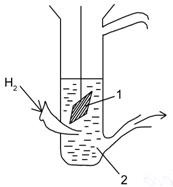 1 – платинированная платина
1 – платинированная платина
2 – концентрированная серная кислота, омывается потоком водорода
Три состояния водорода:
а) молекулярный водород;
б)![]() - молекулы водорода адсорбированные на поверхности
- молекулы водорода адсорбированные на поверхности
платины;
в)![]() - ионы в растворе. Электродные реакции:
- ионы в растворе. Электродные реакции:
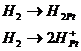
![]()
Суммарная реакция является каталитической и протекает на поверхности платины.
По уравнению Нернста:
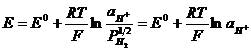 ,
,
где ![]() - стандартный потенциал водородного электрода (нормальный).
- стандартный потенциал водородного электрода (нормальный).
При всех температурах, 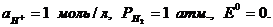
Электродный потенциал любого другого электрода можно определить как ЭДС гальванического элемента, составленного из исследуемого электрода и стандартного водородного. Водородный электрод обозначается в схеме слева.
Пример: Для определения электродного потенциала медного электрода:
![]()
С термодинамической точки зрения самопроизвольному процессу ( убыль (![]() )) соответствует положительная ЭДС : то есть положительный потенциал у медного электрода и отрицательный у водородного. ЭДС этого элемента можно представить как следующую алгебраическую сумму:
)) соответствует положительная ЭДС : то есть положительный потенциал у медного электрода и отрицательный у водородного. ЭДС этого элемента можно представить как следующую алгебраическую сумму:
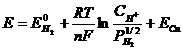 ,
,
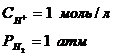
![]() > 0.
> 0.
Для нахождения ЭДС цинкового электрода ![]() (Е < 0):
(Е < 0):
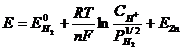 ,
,
![]() .
.
Электроды второго рода – это системы, в которых металл электрода покрыт слоем его малорастворимого соединения и погружен в раствор хорошо растворимой соли, содержащей тот же анион. Сюда относятся хлорсеребряный электрод и каломельный электрод.
Рис. 9 Х лорсеребряный электрод
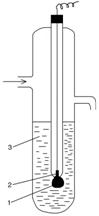 Серебряный электрод (1) , часть поверхности которого электролитически покрыта слоем труднорастворимой соли AgCl (2), погружен в насыщенный раствор AgCl в хлориде KCl (3). На границе раздела металл - раствор протекает окислительно-восстановительная реакция:
Серебряный электрод (1) , часть поверхности которого электролитически покрыта слоем труднорастворимой соли AgCl (2), погружен в насыщенный раствор AgCl в хлориде KCl (3). На границе раздела металл - раствор протекает окислительно-восстановительная реакция:
![]()
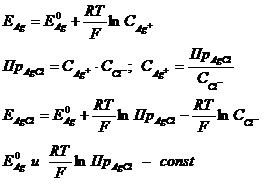 Потенциал такого электрода, согласно уравнению Нернста, определяется концентрацией катионов серебра в растворе:
Потенциал такого электрода, согласно уравнению Нернста, определяется концентрацией катионов серебра в растворе:
при данной температуре Т.
Следовательно, ![]() стандартный потенциал хлорсеребряного электрода, то есть
стандартный потенциал хлорсеребряного электрода, то есть ![]() определяется концентрацией ионов
определяется концентрацией ионов ![]() в растворе. Потенциал хлорсеребряного электрода с насыщенным раствором KCl зависит только от температуры и рассчитывается по формуле:
в растворе. Потенциал хлорсеребряного электрода с насыщенным раствором KCl зависит только от температуры и рассчитывается по формуле:
![]()
Рис.10 Каломельный электрод:
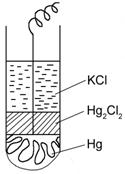 Ртуть покрыта пастой, которая содержит каломель
Ртуть покрыта пастой, которая содержит каломель![]() –слаборастворимое соединение, в состав которого входит анион хлора; электрод помещен в раствор KCl.
–слаборастворимое соединение, в состав которого входит анион хлора; электрод помещен в раствор KCl.
![]()
анод катод
![]()
В зависимости от концентраций KCl, которая является постоянной, различают 1 Н; 0,1 Н; насыщенный каломельный электроды.
Электроды третьего рода – система, которая состоит из металла, который контактирует с двумя труднорастворимыми солями. В результате химической реакции менее растворимая соль превращается в более растворимую.
Свинцовый электрод
Находится в контакте с солями PbCl2 и AgCl.
![]()
Суммарная реакция: 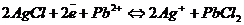 .
.
Окислительно-восстановительные электроды – системы, в которых потенциало-образующие окислительно-восстановительные реакции идут без участия материала самого электрода (идет в растворе электролита без участия самого металла; электроды только осуществляют передачу электронов). Инертны.
Пример:  .
.
![]() .
.
Электрод «+» (катод), если будет идти реакция восстановления.
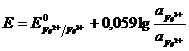
В ряду напряженностей металлов: ![]() .
.
Электрохимические цепи
Химические цепи состоят из электродов, потенциало-образующие химические реакции которых различны и определены равенством: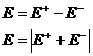 .
.
Подобные цепи реализуются в первичных источниках тока – гальванических элементах, и во вторичных – аккумуляторах.
2. Концентрационные цепи состоят из электродов с одинаковыми потенциало-образующими реакциями, которые отличаются друг от друга активностью растворов. Могут образовываться при контакте двух растворов электролитов с различной активностью ионов, что обусловлено возникновением диффузионного потенциала на границе двух электролитов. В концентрационных электрохимических цепях используют один электролит, но различные электроды (например, амальгамные или газовые).
ЭДС концентрационного элемента позволяет определить некоторые физико-химические свойства растворов: активность, константу активности, числа переноса, растворимость.
Топливные генераторы – электрохимические генераторы, в которых происходит непрерывная подача вещества.
Зависимость ЭДС от температуры
В случае самопроизвольного процесса: ![]() < 0, Emax > 0. Из (2.17):
< 0, Emax > 0. Из (2.17):
![]() ,
,
где ![]() – теплота при обратимом процессе,
– теплота при обратимом процессе, ![]() – теплота, когда не совершается полезная работа.
– теплота, когда не совершается полезная работа.
![]()
![]()
![]()
![]() , где
, где ![]() температурный коэффициент.
температурный коэффициент.
Тепловой эффект гальванического элемента: ![]() (3.10)
(3.10)
Х и м и ч е с к о е р а в н о в е с и е.
Общее условия химического равновесия.
Изобарный потенциал гомогенной многокомпонентной системы представляет собой однородную функцию первого порядка по числу молей компонентов:
G = G (T,p,n1,n2,…). (4.1)
GT,p= 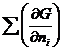 T,p,n
T,p,n![]() dni =
dni =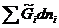 =
=![]() ,
,
где ![]() =
=![]() =
=![]() T,p,n
T,p,n![]() - (4.2)
- (4.2)
Химический потенциал i-го компонента равен приращению изобарного потенциала при добавлении одного моля этого компонента к большому объему системы при постоянных температуре и давлении и постоянных количествах других компонентов.
Рассмотрим находящуюся в неизменных внешних условиях термодинамическую систему, в которой самопроизвольно протекает химическая реакция:
![]()
![]() +
+![]()
![]() + …=
+ …= ![]()
![]() +
+![]()
![]() + … ( 4.3)
+ … ( 4.3)
Сразу после контакта исходных веществ в системе образуются продукты прямой реакции, которые, в свою очередь, вступают в химическое взаимодействие с образованием исходных веществ (обратная реакция). По мере протекания самопроизвольного процесса число молекул исходных веществ, реагирующих в единицу времени (скорость прямой реакции) снижается, так как концентрации исходных веществ уменьшаются, а скорость обратной реакции увеличивается. Когда скорости прямой и обратной реакции сравняются, наступает состояние химического равновесия, характеризуемое постоянством числа молекул исходных и конечных веществ. В состоянии химического равновесия число молекул исходных веществ, вступающих в единицу времени в прямую реакцию, равно числу молекул конечных веществ, участвующих в обратном процессе за тот же промежуток времени.
С изменением внешних условий химическое равновесие нарушается, что вызывает изменение концентраций реагирующих веществ и всех термодинамических функций, характеризующих состояние системы. С течением времени система вновь придет в состояние равновесия, которое будет характеризоваться минимальным в данных условиях значением соответствующего термодинамического потенциала.
Чаще всего химические реакции протекают в условиях постоянства давления и температуры. В этом случае при достижении равновесия изобарно-изотермический потенциал принимает минимальное значение:
dG = 0 (4.4)
Полный дифференциал изобарно-изотермического потенциала гомогенной многокомпонентной системы имеет вид
dG = - SdT + Vdp +![]() (4.5)
(4.5)
Учитывая, что все вещества системы участвуют в химической реакции, изменение числа молей можно представить как
![]() =
=![]() , (4.6)
, (4.6)
где ![]() – химическая переменная.
– химическая переменная.
Тогда уравнение (4.5) принимает форму:
dG = - SdT + Vdp + ![]() (4.7)
(4.7)
Из выражения (4.7) следует, что при протекании в системе одной химической реакции, изобарно-изотермический потенциал является функцией трех переменных:
G = G ( T,p,![]() ) (4.8)
) (4.8)
и, следовательно,
dG = 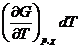 +
+ 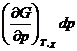 +
+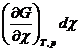 (4.9)
(4.9)
Из уравнения (4.7) и (4.9) следует, что
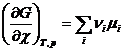 (4.10)
(4.10)
Так как химическая реакция протекает при условии постоянства давления и температуры, то
dG = =![]() (4.11)
(4.11)
В состоянии химического равновесия выполняется уравнение (4.4) и из (4.11) следует, что
![]() = 0 и
= 0 и 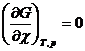 (4.12)
(4.12)
Уравнения (4.11) и (4.12) выражают в общем виде условие химического равновесия.
![]() (р) =
(р) =![]() + RT lnрί ( 4.13 )
+ RT lnрί ( 4.13 )
![]() (с) =
(с) =![]() + RT lnсί (4.14)
+ RT lnсί (4.14)
Для реальных газов уравнения (4.13) и ( 4.14). являются лишь некоторым приближением, работающим при низких давлениях. Для сохранения формы уравнения давление (![]() ) заменяют другой величиной – летучестью или фугитивностью ( f ). Летучестью называется такое давление реального газа, при котором газ ведет себя как идеальный (величина имеет размерность давления):
) заменяют другой величиной – летучестью или фугитивностью ( f ). Летучестью называется такое давление реального газа, при котором газ ведет себя как идеальный (величина имеет размерность давления):
![]() (f) =
(f) =![]() + RT ln fί (4.15)
+ RT ln fί (4.15)
Чтобы найти летучесть, обычно ее выражают через p
f = ![]()
 p, где
p, где ![]() – безразмерная величина, называемая коэффициентом летучести. (
– безразмерная величина, называемая коэффициентом летучести. ( ![]() =
= ![]() ( T, p )).
( T, p )).
Перепишем (4.15):
![]() (f) =
(f) = ![]() +RT ln
+RT ln![]() + RT ln pί (4.16)
+ RT ln pί (4.16)
Таким образом, летучесть (фугитивность) – величина, которую надо подставить в выражение для химического потенциала идеального газа, чтобы получить значение химического потенциала реального газа. Аналогично для растворов, где мерой количества вещества является молярная концентрация:
p=![]() RT
RT![]() =CRT
=CRT
![]() (C2) =
(C2) = ![]() (C1) + RT ln
(C1) + RT ln![]() , С1 = 1 моль.
, С1 = 1 моль.
для идеальных: ![]() (Сi) =
(Сi) =![]() + RT ln Сί
+ RT ln Сί
для реальных: ![]() (Сi) =
(Сi) =![]() + RTln
+ RTln![]() + RT ln αί (4.17)
+ RT ln αί (4.17)
aί – активность компонента ί ;
![]() – коэффициент активности компонента ί.
– коэффициент активности компонента ί.
Равновесие в реакциях, протекающих в газовой фазе.
Общее условие равновесия системы можно записать так:
∆G = 0 или ∑ ![]() dnί = 0 , другими словами
dnί = 0 , другими словами
условие равновесия – равенство нулю суммы мольных значений свободной энергии Гиббса (или химических потенциалов).
Для реакции
aA + bB = cC + dD
∆G = ∑∆Gпрод – ∑∆Gисх = (c∆GC + d∆GD – b∆GB – a∆GA), но
![]() (p) =
(p) =![]() + RT ln pί ,
+ RT ln pί ,
Следовательно, ∆G = c![]() + d
+ d![]() – b
– b![]() – a
– a![]() = c
= c![]() + d
+ d![]() – b
– b![]() – a
– a![]() +
+
+ RT(c ln pC + d ln pD – b ln pB – a ln pA) =
= ∆Gо + RT ln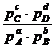 (4.18)
(4.18)
Мы получили изотерму химической реакции или изотерму Вант-Гоффа, где под знаком логарифма ставится константа равновесия (![]() ) для химической реакции между газами.
) для химической реакции между газами.
В условиях равновесия ∆G0 + RT ln![]() = 0, где
= 0, где ![]() =
= ![]() ,
,
ln![]() = –
= –![]() (4.19)
(4.19)
Таким образом, ![]() можно рассчитать из величины энергии Гиббса,
можно рассчитать из величины энергии Гиббса, ![]() в этом случае выражается в единицах давления в степени ∆n (в атмосферах или Па).
в этом случае выражается в единицах давления в степени ∆n (в атмосферах или Па).
Равновесие в реакциях, протекающих в растворах.
Воспользовавшись (4.18), получим
∆G0 + RT ln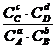 = 0 (4.20),
= 0 (4.20),
откуда ![]() =
= ![]() = .
= .
Или более применимая для термодинамических расчетов реальных растворов константа ![]() =
= ![]() =
= 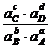
Рассмотрим связь между ![]() и
и![]() :
:
ai =![]() Ci
Ci
![]() =
= ![]() =
=
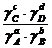 =
=![]() (4.21)
(4.21)
![]() и
и![]() :
:
pV = ![]() nRT; p =
nRT; p =![]() ;
; ![]() = C (молярная концентрация), следовательно
= C (молярная концентрация), следовательно
p = СRT
![]() =
=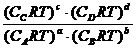 = (RT)c+d-b-a
= (RT)c+d-b-a
![]() = (RT)
= (RT)![]() n
n ![]()
![]() – величина, позволяющая количественно выразить химическое равновесие и условия, определяющие выход продуктов реакции.
– величина, позволяющая количественно выразить химическое равновесие и условия, определяющие выход продуктов реакции.
Равновесие в гетерогенных системах.
Если система находится в состоянии равновесия, то должно выполняться условие равенства всех интенсивных параметров во всех присутствующих фазах. Это справедливо и для химических потенциалов. Таким образом, для реакции
BaSO4 ↔ Ba2+ (раствор) + SO42- (раствор)
Химический потенциал иона Ba2+ (![]() Ba2+) в насыщенном растворе BaSO4 равен
Ba2+) в насыщенном растворе BaSO4 равен ![]() Ba2+ в осадке. Хотя реальное различие в концентрациях может достигать десяти и более порядков.
Ba2+ в осадке. Хотя реальное различие в концентрациях может достигать десяти и более порядков.
Рассмотрим процесс разложения CaCO3.
CaCO3 = CaO + CO2 ↑
CaO и CO2 не образуют твердых растворов. Таким образом, концентрация этих веществ в области протекания реакции равны, а, следовательно, и их химические потенциалы не будут меняться вплоть до полного исчезновения одного из компонентов и будут равны соответствующим стандартным величинам, а для CO2:
![]() =
= ![]() + RT ln
+ RT ln![]() ,
,
отсюда при равновесии при атмосферном давлении:
∆G = ![]() + µCaO –
+ µCaO –![]() =
=![]() + RT ln
+ RT ln![]() +
+![]() –
–![]() = ∆G0 CaCO3 + RT ln
= ∆G0 CaCO3 + RT ln![]() или
или
![]() =
=![]()
Таким образом, ![]() в этом случае является парциальным давлением CO2. В общем случае для получения
в этом случае является парциальным давлением CO2. В общем случае для получения ![]() гетерогенного процесса с участием газов следует учитывать лишь парциальное давление газов. Аналогичное заключение и для реакций в растворах, сопровождающихся выпадением или растворением осадка.
гетерогенного процесса с участием газов следует учитывать лишь парциальное давление газов. Аналогичное заключение и для реакций в растворах, сопровождающихся выпадением или растворением осадка.
Ф а з о в ы е р а в н о в е с и я.
Правило Фаз Гиббса.
Предположим, что гетерогенная система состоит из ф фаз, в каждую из которых входят к компонентов. В дальнейшем нижними индексами будем обозначать компоненты системы, а верхними фазы (так как это сделано в выражении (5.1) для обозначения химических потенциалов).
При равновесии многокомпонентной гетерогенной системы должны выполняться следующие условия.
а) условие термического равновесия:
T1 = T2 = … = Tф (5.2)
б) условие механического равновесия:
p1 = p2 = … = pф (5.3)
в) условие химического равновесия:
![]() =
= ![]() = … =
= … = ![]()
![]() =
= ![]() = … =
= … =![]() (5.4)
(5.4)
![]() =
= ![]() = … =
= … = ![]()
Выражения (5.2) и (5.3) представляют собой ряды тождеств, так как температура и давление являются независимыми переменными. Химические потенциалы представляют собой функции, зависящие от температуры, давления и концентраций. Общий вид этих функций может быть выражен, например, таким образом:
![]() =
= ![]() ( p, T, x1, x2, … xк) (5.5)
( p, T, x1, x2, … xк) (5.5)
где x1, x2, …, xк - концентрации.
При переходе от одной фазы к другой вид функции (5.5) меняется, поэтому равенства типа
![]() =
= ![]() ;
; ![]() =
= ![]() и т.д. представляют собой уравнения.
и т.д. представляют собой уравнения.
Каждая cтрока системы (5.4) позволяет составить ( ф - 1 ) независимых уравнений. Число cтрок в системе равно к, поэтому общее число независимых уравнений
к ( ф - 1 ) (5.6)
В число независимых переменных входят температура, давление и концентрации компонентов. В каждой фазе к компонентов, но при заданных температуре и давлении выбрать произвольно к концентрации нельзя. Например, для смеси не реагирующих между собой идеальных газов согласно закону Дальтона:
p = p1 + p2 + … + pк
При заданном общем давлении p можно произвольно менять парциальные давления кроме одного. Следовательно, независимых переменных в этой случае (к - 1 ).
Другой пример: жидкий раствор, в состав которого входят к компонентов с концентрациями x1, x2, … xк, выраженные в мольных долях. Ясно, что x1 + x2 + … + xк = 1, и, следовательно, при произвольной изменении (к - 1) концентрации, выбор последней концентрации определится уравнением ![]() = 1.
= 1.
Для каждой фазы можно записать уравнение состояния, которое связывает температуру, давление и концентрации компонентов, поэтому число независимых концентраций в каждой фазе (к - 1). Общее число независимых переменных с учетом температуры и давления будет:
ф(к - 1) + 2 (5.7)
Если число независимых переменных (5.7) больше числа независимых уравнений (5.6), то разность этих двух чисел
С = ф(к - 1) + 2 - к( ф - 1 )
представляет собой число переменных, которым можно придавать произвольные значения при данном числе фаз.
С – число степеней свободы
С = к + 2 - ф (5.8)
Соотношение (5.8) называется законом фазового равновесия или правилом фаз Гиббса.
Число степеней свободы - число независимых переменных (давление, температура, концентрации), которые можно менять в некоторых пределах без изменения числа фаз. Система, имеющая две степени свободы, называется бивариантной, а имеющая одну степень свободы - моновариантной. Если числи степеней свободы равно нулю, то такая система инвариантна.
Фазовое равновесие в однокомпонентных системах.
Уравнение Клапейрона-Клаузиуса
Рассмотрим закономерности, связанные с превращением одной фазы чистого вещества в другую: испарение, плавление, переход твердого тела из одной модификации в другую. Для примера возьмем равновесие между жидкостью и её паром при постоянных температуре и давлении. Для этого поместим жидкость на дно цилиндра, погруженного в термостат. Давление пара, находящегося в равновесии с жидкостью, уравновешено внешним давлением ( Рис.10 ).
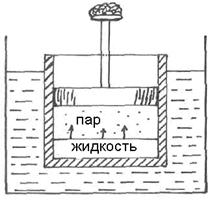
Рис.10
Условие равновесия такой системы согласно (4.12):
![]()
В силу постоянства состава в однокомпонентной системе химический потенциал тождественен мольному термодинамическому потенциалу (![]() ).
).
Следовательно, условие равновесия может быть записано так:
![]() . (5.9)
. (5.9)
При повышении давления на бесконечно малую величину dp мольные изобарно-изотермические потенциалы жидкости и пара изменяются также на бесконечно малую величину:
dG = - SdT + Vdp
При Т =const для одного моля жидкости и пара соответственно получим:
![]()
![]()
Так как мольный объем пара (![]() ) больше мольного объема жидкости (
) больше мольного объема жидкости (![]() ) , то
) , то ![]() при установлении нового состояния равновесия часть пара должна превратиться в жидкость. Этот вывод согласуется с принципом Ле Шателье-Брауна, согласно которому под воздействием внешних сил система должна так изменить свое состояние, чтобы уменьшить это воздействие. Если при постоянном давлении повысим температуру на dТ, то изменение изобарно-изотермических потенциалов жидкости и пара будет иметь вид:
при установлении нового состояния равновесия часть пара должна превратиться в жидкость. Этот вывод согласуется с принципом Ле Шателье-Брауна, согласно которому под воздействием внешних сил система должна так изменить свое состояние, чтобы уменьшить это воздействие. Если при постоянном давлении повысим температуру на dТ, то изменение изобарно-изотермических потенциалов жидкости и пара будет иметь вид:
![]()
![]()
Ясно, что 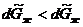 ,так как мольная энтропия пара (
,так как мольная энтропия пара (![]() ) больше мольной энтропии жидкости (
) больше мольной энтропии жидкости (![]() ). Следовательно, при установлении нового состояния равновесия часть жидкости должна будет испаряться. Это также подтверждается принципом Ле Шателье-Брауна, так как при повышении температуры в системе должны идти процессы с поглощением тепла.
). Следовательно, при установлении нового состояния равновесия часть жидкости должна будет испаряться. Это также подтверждается принципом Ле Шателье-Брауна, так как при повышении температуры в системе должны идти процессы с поглощением тепла.
В самом общем случае при постоянных температуре и давлении равновесие двух фаз однокомпонентной системы:
1 фаза![]() 2 фаза
2 фаза
можно записать, как ![]()
После одновременного изменения температуры на dT и давления на dp установится новое состояние равновесия:
![]()
![]()
![]()
Запишем уравнения полных дифференциалов для изобарно-изотермических потенциалов 1 и 2 фаз:
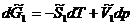
![]()
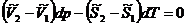
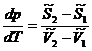 (5.10)
(5.10)
Для разновесных изотермических фазовых переходов
![]()
(5.11)
где ![]() – мольная теплота фазового перехода,
– мольная теплота фазового перехода,
![]() – температура фазового перехода.
– температура фазового перехода.
Подставим (5.11) в уравнение (5.10), получим
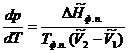 (5.12)
(5.12)
Уравнение (5.12) называется уравнением Клапейрона-Клаузиуса.
Фазовые переходы 1 рода.
Фазовые переходы, характеризующиеся равенством изобарно-изотермических потенциалов равновесных фаз и скачкообразным изменением энтропии и объема при переходе вещества из одной фазы в другую, называется фазовыми переходами первого рода.
Энтропия и объем являются первыми производными изобарно-изотермического потенциала по соответствующим переменным ( см. раздел Термодинамика):
![]() ;
; 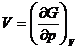
Поэтому фазовые переходы 1 рода характеризуются скачкообразным изменением первых производных изобарного потенциала.
Испарение - пример фазового перехода 1 рода:
Переход жидкой фазы в газообразную сопровождается поглощением тепла (![]() ). Мольный объем газа всегда больше соответствующего объема жидкости
). Мольный объем газа всегда больше соответствующего объема жидкости ![]() Поэтому при фазовом переходе
Поэтому при фазовом переходе ![]() в уравнении Клапейрона-Клаузиуса. (5.12) производная
в уравнении Клапейрона-Клаузиуса. (5.12) производная ![]() всегда положительна. Следовательно, температура испарения всегда повышается с ростом давления.
всегда положительна. Следовательно, температура испарения всегда повышается с ростом давления.
При температурах, далеких от критической, мольной объем пера всегда во много раз превосходит мольный объем жидкости. Поэтому можно пренебречь мольным объемом жидкости в уравнении (5.12), которое принимает вид:
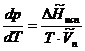 (5.13)
(5.13)
Если пар вдали от критической точки считать идеальным газом, то
![]() (5.14)
(5.14)
Подставим (5.14) в уравнение (5.13) и проинтегрируем полученное выражение, считая ![]() постоянной величиной в небольшом интервале температур:
постоянной величиной в небольшом интервале температур:
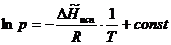
Представив зависимость давления насыщенного пара от температуры в координатах 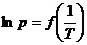 , можно найти мольную теплоту испарения (Рис.11).
, можно найти мольную теплоту испарения (Рис.11).
|
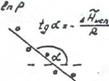
В действительности теплота испарения в области температур далекой от критической, немного уменьшается с ростом температуры и сильно убывает вблизи критической точки, при которой ![]() .Аналогичным уравнением описывается сублимация твердого тела.
.Аналогичным уравнением описывается сублимация твердого тела.
Теплоты испарения различных жидкостей связаны с температурами кипения эмпирическим правилом Трутона: изменение мольной энтропии испарения различных жидкостей при нормальной температуре кипения одинаковы:
![]()
(5.15)
Это правило не выполняется для ассоциированных и полярных жидкостей.
Плавление является также примером фазового перехода 1 рода и к нему применимо уравнение Клапейрона-Клаузиуса:
тв. тело![]() жидкость
жидкость
(I) (П)
Для этого процесса
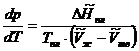 (5.16)
(5.16)
Теплота плавления - теплота перехода твердой фазы в жидкую – положительна 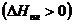 . Мольные объемы твердого тела и жидкости сравнимы между собой. Для большинства веществ мольный объем жидкости больше мольного объема твердого тела (
. Мольные объемы твердого тела и жидкости сравнимы между собой. Для большинства веществ мольный объем жидкости больше мольного объема твердого тела (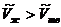 ). В этом случае из уравнения (5.16) следует, что производная
). В этом случае из уравнения (5.16) следует, что производная ![]() положительна, а это значит, что с повышением давления увеличивается температура плавления. Для таких веществ, как вода, висмут и некоторых других при температуре плавления мольный объем жидкости меньше мольного объема твердого тела (
положительна, а это значит, что с повышением давления увеличивается температура плавления. Для таких веществ, как вода, висмут и некоторых других при температуре плавления мольный объем жидкости меньше мольного объема твердого тела (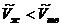 ). Следовательно,
). Следовательно,![]() < 0 и с повышением давления температура плавления указанных веществ понижается.
< 0 и с повышением давления температура плавления указанных веществ понижается.
Фазовые переходы 2 рода
Фазовые переходы, при которых не изменяются не только изобарно-изотермический потенциал (ΔG = 0), но и его первые производные (ΔV = 0, ΔS = 0),а скачкообразно изменяются вторые производные :
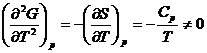 (5.17)
(5.17)
называются фазовыми переходами второго рода.
Фазовые переходы второго рода не сопровождаются тепловыми эффектами. К фазовым переходам второго рода относятся переходы металла в сверхпроводящее состояние, ферромагнитных веществ в парамагнитные, изменения типа симметрии кристаллов. Как видно из выражений (5.17), о наличии фазовых переходов второго рода можно судить по скачкообразным изменениям на кривых, характеризующих температурные зависимости теплоемкости и мольного объема.
Диаграмма состояния воды.
Графическое изображение зависимости состояния системы и фазовых равновесий в ней от внешних условий и от её состава называется диаграммой состояния или фазовой диаграммой.
|
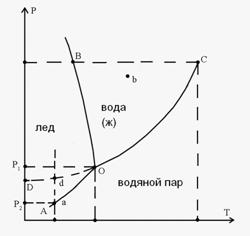
Рис.12 Диаграмма состояния воды.
На Рис. 12 приведена диаграмма воды в области средних давлений. Область диаграммы левее кривой AОB соответствует твердому состоянию (лёд). Область диаграммы ниже кривой АОС соответствует газообразному состоянию воды. Часть диаграммы, заключенная между кривыми ОВ и ОС, отвечает жидкому состоянию. Кривые ОА, ОВ, ОС соответствуют равновесию между соответствующими двумя фазами. Кривая ОА выражает зависимость давления насыщенного пара надо льдом от температуры. Кривая ОВ изображает зависимость температуры плавления льда от давления. Наклон кривой OB влево свидетельствует об отрицательной значении производной ![]() , т.е. температура замерзания понижается с повышением давления.
, т.е. температура замерзания понижается с повышением давления.
Кривая ОС характеризует зависимость давления насыщенного пара жидкой воды от температуры. Все рассмотренные кривые начинаются в точке О, которая соответствуют сосуществованию трех агрегатных состояний воды: жидкого, твердого и газообразного. Эта точка называется тройной.
Рассчитать число степеней свободы в точке О, можно воспользоваться правилом фаз Гиббса (5.8). Рассматривая система однокомпонентная ( к = 1) и в данной точке Ф = 3, так как три фазы находятся в равновесии. С = к + 2 – ф = 1 + 2 – 3 = 0, что означает, что указанному состоянию соответствует строго определенные значения Т и р: 273,16 К и 0,006 атм. Точка а, лежащая на кривой АО, соответствует равновесию двух фаз: льда и пара. Число степеней свободы в этой точке С = 1 + 2 – 2 = 1, что означает, что изменение одного параметра (например температуры) повлечет за собой изменение другого (давления) при сохранении равновесия твердой и газообразной фазы. Уравнение Клапейрона-Клаузиуса (5.12) позволяет рассчитать значение давления при заданном изменении температуры.
Число степеней свободы в точке в равно 2, так как в этой точке всего одна фаза:
С = I + 2 – 1 = 2. Это означает, что в определенных интервалах можно менять температуру и давление при сохранении жидкой фазы. Пределы изменения температуры и давления определяются кривыми OB и ОС.
Однако при осторожном охлаждении воды можно получить её в переохлажденном состоянии. Пунктирная кривая ОД отвечает давлению насыщенного пара над переохлажденной жидкостью. Такое состояние неустойчиво и носит название метастабильного. Кривая ОД расположена выше кривой ОА, которая характеризует зависимость давления насыщенного пара надо льдом. При попадании в сосуд с переохлажденной водой твердых частиц или при легком ударе о сосуд переохлажденная вода переходит в лёд без изменения температуры. Возможность такого самопроизвольного перехода вытекает из следующих рассуждений. При постоянной температуре ![]() . Считая, что при низких давлениях пар представляет собой идеальный газ, получим
. Считая, что при низких давлениях пар представляет собой идеальный газ, получим
![]()
Проинтегрировав последнее выражение, имеем:
![]()
(5.18)
Как видно из Рис. 12, р2 < р1, а следовательно в уравнении (5.18) ΔG <О. Таким образом, переход из метастабильного состояния, представляющего собой переохлажденную жидкость с давлением насыщенного пара p1 (точка ![]() ), в устойчивое твердое состояние с давлением пара p2 (точка а ) самопроизвольный.
), в устойчивое твердое состояние с давлением пара p2 (точка а ) самопроизвольный.
Равновесие бинарного жидкого раствора, состоящего из летучих веществ,с паром.
Пар, находящийся в равновесии в равновесии с жидкостью, называется насыщенным паром. Жидкости, которые при данной температуре имеют достаточно высокое давление пара, называются летучими.
В самом общем случае жидкий раствор, представляющий собой смесь летучих жидкостей, находится в равновесии с паром, состоящим из тех же компонентов, что и жидкая фаза. Давление насыщенного пара над жидким раствором будет определяться суммой парциальных давлений компонентов. Однако, когда в состав жидкой фазы входят нелетучие вещества, последние могут отсутствовать в газовой фазе.
Рассмотрим раствор двух летучих жидкостей А и В. Мольную долю компонента В обозначим x, тогда мольная доля компонента А будет равна (1-х). При постоянной температуре давление насыщенного пара будет функцией только состава раствора. На диаграмме, представленной на рис 13, изображена зависимость давления насыщенного пара раствора от его состава. По оси абсцисс слева направо обычно откладывают мольную долю компонента В, имеющего в чистом виде более высокое давление насыщенного пара и поэтому более низкую температуру кипения. Мольную долю компонента А легко вычислить, учитывая, что сумма мольных долей обоих компонентов, равны единице.
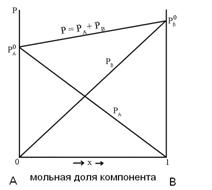
Рис. 13
Из рис.13 видно, что давление насыщенного пара каждого из компонентов пропорционально его мольной доле в растворе, а коэффициентом пропорциональности является давление насыщенного пара чистого вещества.
![]() (5.19)
(5.19)
![]() (5.20)
(5.20)
Приведенные соотношения, полученные экспериментально, носят название закона Рауля. Выражение (5.19) легко преобразовать:
![]() (5.21)
(5.21)
Относительное понижение давление пара одного компонента бинарного раствора равно мольной доле второго компонента в растворе.
Растворы, подчиняющиеся закону Рауля, называются идеальными.
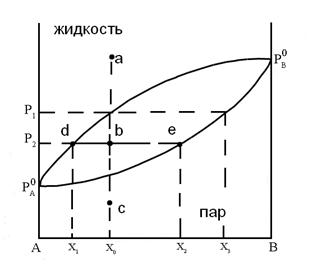
Рис.14 Диаграмма состояния бинарного жидкого
раствора и равновесного с ним пара при постоянной температуре.
Любая точка на диаграмме, называемая фигуративной, характеризует состав системы и давления. Выше кривой ![]() лежит однофазная область - бинарный жидкий раствор, который обладает двумя степенями свободы. Это следует из уравнения (5.8), примененного к точке "а"
лежит однофазная область - бинарный жидкий раствор, который обладает двумя степенями свободы. Это следует из уравнения (5.8), примененного к точке "а"
![]()
Это значит, что для однозначного определения состояния системы с одной фазой нужно задать мольную долю компонента и давление. Бивариантной система будет также в области ниже кривой ![]() , соответствующей однофазной газовой смеси переменного состава.
, соответствующей однофазной газовой смеси переменного состава.
поле диаграммы, заключенное между кривыми ![]() и
и ![]() , соответствует двухфазной системе: жидкий раствор и равновесный с ним пар. Верхняя кривая
, соответствует двухфазной системе: жидкий раствор и равновесный с ним пар. Верхняя кривая ![]() характеризует зависимость давления насыщенного пара от состава жидкости и называется линией жидкости (ликвидуса), а кривая
характеризует зависимость давления насыщенного пара от состава жидкости и называется линией жидкости (ликвидуса), а кривая ![]() – зависимость давления насыщенного пара от состава пара. Это- линия пара. Любая фигуративная точка, заключенная между ветвями жидкости и пара, отвечает системе с одной степенью свободы. Если через точку «b» проведем изобару, то точки ее пересечения с ветвями жидкости (a) и пара (c) будут характеризовать равновесные составы жидкости и пара соответственно. Такие точки, выражающие составы двух равновесных фаз при заданных T и p, называются сопряженными точками, а линии, соединяющие сопряженные точки (dl), называются нодами, или коннодами. Состав пара отличается от состава равновесной с ним жидкости. При изотермическом сжатии ненасыщенного пара состава x2 фигуративная точка поднимается вверх. В точке «c» пар становится насыщенным, появляются первые капли жидкости состава x1, то есть жидкость обогащена компонентом А по сравнению с равновесным с ней паром. При изотермическом уменьшении давления точка «а», характеризующая жидкую фазу, смещается вниз и при давлении
– зависимость давления насыщенного пара от состава пара. Это- линия пара. Любая фигуративная точка, заключенная между ветвями жидкости и пара, отвечает системе с одной степенью свободы. Если через точку «b» проведем изобару, то точки ее пересечения с ветвями жидкости (a) и пара (c) будут характеризовать равновесные составы жидкости и пара соответственно. Такие точки, выражающие составы двух равновесных фаз при заданных T и p, называются сопряженными точками, а линии, соединяющие сопряженные точки (dl), называются нодами, или коннодами. Состав пара отличается от состава равновесной с ним жидкости. При изотермическом сжатии ненасыщенного пара состава x2 фигуративная точка поднимается вверх. В точке «c» пар становится насыщенным, появляются первые капли жидкости состава x1, то есть жидкость обогащена компонентом А по сравнению с равновесным с ней паром. При изотермическом уменьшении давления точка «а», характеризующая жидкую фазу, смещается вниз и при давлении ![]() образуется пар состава x3 , обогащенный компонентом B по сравнению с жидкой фазой.
образуется пар состава x3 , обогащенный компонентом B по сравнению с жидкой фазой.
Отклонения от закона Рауля.
Для многих систем из двух летучих жидкостей закон Рауля не выполняется. Отклонения давления пара от линейной зависимости в сторону больших значений называют положительными, а в сторону меньших значений – отрицательными. Причина положительных отклонений от закона Рауля заключается в том, что разнородные молекулы притягиваются слабее однородных. При образовании жидкого раствора межмолекулярное взаимодействие ослабляется, и возможность перехода молекул в парообразное состояние увеличивается. При этом давление насыщенного пара будет больше, чем следовало бы по закону Рауля. Отрицательные отклонения от закона Рауля наблюдаются в том случае, когда взаимодействие между разнородными молекулами больше, чем между однородными. Это вызывает уменьшение давления насыщенного пара по сравнению с величиной, наблюдаемой для идеальных растворов.
Положительные отклонения от закона Рауля наблюдаются в системах ацетон-этанол, вода-метанол, четыреххлористый углерод-бензол, четыреххлористый углерод-хлороформ, а отрицательные отклонения от закона Рауля у растворов: вода - азотная кислота, ацетон-хлороформ, бензол-хлороформ.
Законы Вревского.
Первый закон Вревского показывает, как влияет изменение температуры на состав одной из фаз при заданной составе другой фазы: при повышении температуры раствора заданного состава его пар обогащается тем компонентом, парциальная молярная теплота испарения которого больше. Этот закон справедлив как для растворов с азеотропом, так и без него. Азеотропным называется раствор, у которого состав пара совпадает с составом жидкости. На рис. 15 показано влияние температуры на состав пара при заданном составе жидкости (без азеотропа). Теплота испарения труднолетучего вещества больше теплоты испарения легколетучего, поэтому равновесный с жидкостью пар с повышением температуры будет обогащаться компонентом А, например, от его мольной доли ![]() до
до ![]() .
.
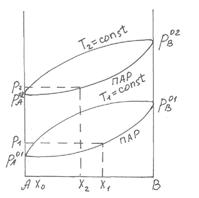
Рис. 15.
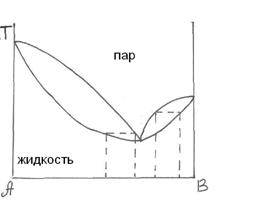 Второй закон Вревского относится только к растворам, образующим азеотроп, и указывает направление изменения состава азеотропа с повышением температуры: если на кривой зависимости давления пара от состава раствора имеется максимум, то при повышении температуры в азеотропном растворе возрастает концентрация того компонента, молярная теплота испарения которого больше. Для систем, имеющих минимум на соответствующих кривых, с повышением температуры в азеотропном растворе возрастает концентрация компонента, молярная теплота испарения которого меньше.
Второй закон Вревского относится только к растворам, образующим азеотроп, и указывает направление изменения состава азеотропа с повышением температуры: если на кривой зависимости давления пара от состава раствора имеется максимум, то при повышении температуры в азеотропном растворе возрастает концентрация того компонента, молярная теплота испарения которого больше. Для систем, имеющих минимум на соответствующих кривых, с повышением температуры в азеотропном растворе возрастает концентрация компонента, молярная теплота испарения которого меньше.
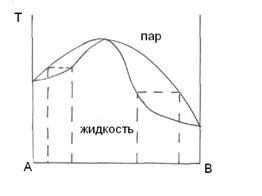
Рис. 16.
Понижение температуры замерзания раствора. Криоскопия.
На рис.17 кривая ОС характеризует давление насыщенного пара над чистым жидким растворителем, а кривая ОА - давление насыщенного пара над твердым растворителем. Точка О характеризует температуру замерзания чистого растворителя. После добавления к растворителю нелетучего вещества его давление насыщенного пара согласно закону Рауля будет во всем интервале температур понижаться, и температурная зависимость давления растворителя будет описываться кривой ![]() при концентрации твердого тела
при концентрации твердого тела ![]() , а при концентрации
, а при концентрации ![]() – кривой
– кривой ![]() . Точки
. Точки ![]() и
и ![]() определяют температуру начала замерзания растворов с соответствующими концентрациями растворенного вещества. Из рис.17 видно, что кристаллизация растворителя начинается при тем более низкой температуре, чем выше в нём концентрация растворенного вещества.
определяют температуру начала замерзания растворов с соответствующими концентрациями растворенного вещества. Из рис.17 видно, что кристаллизация растворителя начинается при тем более низкой температуре, чем выше в нём концентрация растворенного вещества.
![]() , (5.22)
, (5.22)
где![]() – понижение температуры замерзания одного килограмма данного растворителя при добавлении к нему одного моля растворенного вещества.
– понижение температуры замерзания одного килограмма данного растворителя при добавлении к нему одного моля растворенного вещества. ![]() называют молярным понижением точки затвердевания раствора, или криоскопической константой, а изучение температур затвердевания растворов называют криоскопией.
называют молярным понижением точки затвердевания раствора, или криоскопической константой, а изучение температур затвердевания растворов называют криоскопией.
По понижению температуры кристаллизации раствора, зная криоскопическую константу, можно определить молекулярную массу растворенного вещества. Определение молекулярной массы растворенного вещества по изучению температуры замерзания растворов называется криоскопическим методом определения молекулярных масс.
![]() (5.22а)
(5.22а)
Повышение температуры кипения раствора. Эбуллиоскопия.
Рис. 17.
Предположим, что внешнее давление равно одной атмосфере. Тогда, как видно из рис.17, чистый растворитель закипит при температуре ![]() . Тогда как пар раствора нелетучего вещества содержит лишь чистый растворитель, то при данной температуре
. Тогда как пар раствора нелетучего вещества содержит лишь чистый растворитель, то при данной температуре ![]() давление насыщенного пара раствора
давление насыщенного пара раствора ![]() (точка М) будет меньше давления насыщенного пара чистого растворителя
(точка М) будет меньше давления насыщенного пара чистого растворителя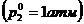 .
.
Пересечение кривой температурной зависимости давления насыщенного пара над раствором с изобарой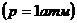 произойдет при температуре
произойдет при температуре ![]() (т. N), которая всегда выше температуры кипения чистого растворителя
(т. N), которая всегда выше температуры кипения чистого растворителя ![]() (Рис.17). Разность
(Рис.17). Разность ![]() будет тем больше, чем больше мольная доля растворенного вещества.
будет тем больше, чем больше мольная доля растворенного вещества.
![]() (5.23)
(5.23)
![]() – мольное повышение температуры кипения, или повышение температуры кипения идеального раствора, состоящего из одного килограмма растворителя и одного моля растворенного вещества. Величина
– мольное повышение температуры кипения, или повышение температуры кипения идеального раствора, состоящего из одного килограмма растворителя и одного моля растворенного вещества. Величина ![]() называется эбуллиоскопической постоянной, она так же как и криоскопическая постоянная зависит только от природы растворителя.
называется эбуллиоскопической постоянной, она так же как и криоскопическая постоянная зависит только от природы растворителя.
Уравнение (3.25) дает возможность вычислить молекулярную массу растворенного вещества, измеряя повышение температуры кипения раствора известной весовой концентрации. Метод определения молекулярной массы по уравнению (5.23а) называется эбуллиоскопией.
![]() (5.23а)
(5.23а)
Осмотическое давление.
Осмос - это самопроизвольное проникновение растворителя в раствор через полупроницаемую мембрану. Осмотическое давление – это избыточное внешнее давление, которое нужно приложить к раствору, чтобы прекратился поток растворителя через полупроницаемую мембрану, отделяющую его от раствора. Рассмотрим в качестве примера систему, состоящую из двух сосудов (Рис.18), один из которых (1) закрыт внизу мембраной 2, через которую может проходить только растворитель. Сосуд 1 с раствором какого-либо вещества помещен в сосуд 3, в котором находится растворитель. Через полупроницаемую мембрану 2 растворитель из сосуда 3 самопроизвольно поступает в сосуд 1, в результате чего там образуется избыточное давление, численно равное осмотическому, но противоположное ему по знаку.
![]() (5.24)
(5.24)
Это выражение носит название уравнение Вант-Гоффа и из него следует, что осмотическое давление разбавленных растворов численно равно давлению, которое производили бы молекулы растворенного в объеме раствора вещества, если бы они в тех же условиях представляли собой идеальный газ.
и из него следует, что осмотическое давление разбавленных растворов численно равно давлению, которое производили бы молекулы растворенного в объеме раствора вещества, если бы они в тех же условиях представляли собой идеальный газ.
