
Партнерка на США и Канаду по недвижимости, выплаты в крипто
- 30% recurring commission
- Выплаты в USDT
- Вывод каждую неделю
- Комиссия до 5 лет за каждого referral
ОЦЕНКА ЭНТАЛЬПИЙ КОМПЛЕКСООБРАЗОВАНИЯ В СМЕШАННЫХ РАСТВОРИТЕЛЯХ НА ОСНОВЕ АДДИТИВНО-ГРУППОВЫХ ВКЛАДОВ
Ивановский государственный химико-технологический университет
При изучении комплексообразования в неводных растворах используется подход, получивший название сольватационно-термодинамического, в котором влияние растворителя на реакцию рассматривается через термодинамические функции DtrY сольватации (или переноса) реагентов [1].
DrY(aq)
Maq + Laq ![]() ® [ML]aq
® [ML]aq
¯ DtrYM ¯ DtrYL ¯ DtrY[ML] (1)
DrY(sol)
Msol + Lsol ![]()
![]() ® [ML]sol
® [ML]sol
На основе термодинамического цикла переноса реагентов (1) можно получить соотношения для устойчивости комплекса и энтальпии реакции комплексообразования в неводном растворителе:
2,3RT( lgKsolv -lgKH2O) = -DtrG°r = DtrG°(L) + DtrG°(M) - DtrG° (ML) (2)
DtrH°r = DrH°sol - DrH°H2O = DtrH°(ML) - DtrH°(L) - DtrH°(M) (3).
В уравнениях (2, 3) величины DtrG° и DtrH° - изменения энергии Гиббса и энтальпии при переносе реагентов ML, L, M из воды в неводный растворитель.
Достоинством сольватационно-термодинамического описания является его универсальный характер, основанный на применении строгих методов классической химической термодинамики для анализа закономерностей комплексообразования. Ранее было показано [2], что изменение энтальпии DtrHr° произвольной реакции комплексообразования под воздействием растворителя зависит, главным образом, от энтальпии процесса переноса соответствующего лиганда DtrH°(L) между растворителями S1 и S2. Причем величины энтальпий находятся в следующем эмпирически установленном отношении, называемом "правилом диапазонов":
0< ôDrH°(S1) -DrH°(S2)ô < ôDtrH°(L)ô (4).
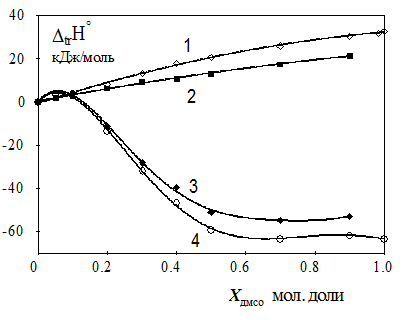 |
Рис. 1. Изменение термодинамических функций для реагентов и реакции образования [NiEn]2+ в смешанном растворителе вода-ДМСО [3]. 1: DtrHo(En) 2: {DrHoводн - DrHoДМСО} 3: DtrHo[NiEn]2+ 4: DtrHo (Ni2+).
Рисунок 1 показывает типичные соотношения между энтальпиями переноса реагентов и изменением экзотермичности реакции. Неравенство (4) хорошо описывает термодинамические параметры реакций, в которых участвуют лиганды, имеющие относительно небольшие размеры молекул: такие как аммиак, алифатические моно- и ди-амины, гетероциклические амины, карбоновые и аминокарбоновые кислоты. Небольшое число изученных систем не подчиняется "правилу диапазонов", например реакция образования никотинамидных комплексов в водно-спиртовых растворителях.
Многочисленные экспериментальные данные доказывают, что между энтальпией сольватации лиганда и изменением энтальпии комплексообразования в неводном растворителе существует корреляция, поскольку наиболее быстрый рост экзотермичности комплексообразования наблюдается, как правило, в условиях, когда происходит наиболее интенсивная десольватация лиганда. При небольших вариациях состава водно-органического растворителя неравенство (5) более точно передает смысл "правила диапазонов":
÷![]() ê ³ ÷
ê ³ ÷![]() ê (5)
ê (5)
где ¶DsolH(L) - изменение энтальпии сольватации лиганда, ¶X2 -изменение состава смешанного растворителя (X2 - мольная доля неводного компонента бинарного растворителя). Таким образом, изменение энтальпии сольватации лиганда всегда больше по величине, чем прирост экзотермичности комплексообразования, вызванный растворителем. При этом предполагается, что изменения в сольватации центрального иона играют в термодинамике комплексообразования менее важную роль, чем сольватация лиганда.
Рассмотрим, каким образом изменение сольватации лиганда влияет на термодинамику комплексообразования в смешанном растворителе. Используем для этого представления [4] об аддитивности вкладов отдельных функциональных групп лиганда в термодинамику процесса сольватации.
Термодинамическую функцию сольватации (переноса) монодентатного лиганда DtrHL можно представить как сумму двух составляющих:
DtrHL = DtrHR + DtrHy (6)
DtrHy - вклад сольватации донорного атома,
DtrHR - вклад сольватации углеводородного фрагмента молекулы лиганда.
Близкое по форме уравнение было использовано в работах [6-7] для определения вкладов углеводородных радикалов и полярных групп в энтальпию сольватации молекулы неэлектролита.
Функцию сольватации (переноса) иона металла DtrHM , имеющего в ближайшем окружении n молекул растворителя, можно представить как сумму вкладов, отражающих сольватацию отдельных координационных мест (электронных орбиталей) центрального иона:
DtrHM = ![]() DtrHM +
DtrHM + ![]() DtrHM (7).
DtrHM (7).
Уравнение (7) не учитывает взаимодействие электрически заряженного сольватокомплекса с растворителем, как со средой, характеризуемой диэлектрической проницаемостью e. Вклад ион-дипольного взаимодействия, вызванный различиями диэлектрических свойств растворителей и рассчитанный по уравнению Борна-Бьеррума, оказывается небольшим по отношению к величине молярной энтальпии переноса иона DtrHM [8,9]. В этой связи уместно привести замечание : "Первичную сольватацию ионов следует рассматривать как процесс комплексообразования. ... Небольшие различия в энергии сольватации изоэлектронных ионов в различных растворителях являются результатом различий в энергии вторичной сольватации". [10].
Энтальпию пересольватация комплексного иона можно представить в виде суммы двух вкладов:
а) вклада тех координационных мест иона, которые не заняты донорным атомом лиганда (![]() DtrHM),
DtrHM),
б) вклада углеводородного фрагмента молекулы лиганда (DtrHR):
DtrHML = ![]() DtrHM + DtrHR (8).
DtrHM + DtrHR (8).
В первом приближении можно принять, что донорный атом лиганда в составе комплекса и занятое им координационное место не участвуют во взаимодействии с растворителем. Тогда из уравнений (3, 6, 7, 8) следует:
DtrHr = - DtrHy - ![]() DtrHM (9).
DtrHM (9).
Таким образом, энтальпия реакции определяется изменениями в сольватном окружении донорного атома лиганда, которые характеризуются величиной DtrHy и сольватационного вклада координационной ячейки центрального иона (-![]() DtrHM) . Последний составляет, по-видимому, небольшую часть от энтальпии переноса иона металла DtrHM, поскольку замена одной или нескольких молекул растворителя, входящих в первую сольватную сферу, на молекулу на лиганда мало сказывается на сольватном состоянии иона металла, имеющего в своем ближнем и дальнем окружении большое число молекул растворителя. Это обстоятельство можно подтвердить термодинамическими параметрами пересольватации ионов Ni2+ и [NiEn]2+ (рис.1), которые мало различаются между собой.
DtrHM) . Последний составляет, по-видимому, небольшую часть от энтальпии переноса иона металла DtrHM, поскольку замена одной или нескольких молекул растворителя, входящих в первую сольватную сферу, на молекулу на лиганда мало сказывается на сольватном состоянии иона металла, имеющего в своем ближнем и дальнем окружении большое число молекул растворителя. Это обстоятельство можно подтвердить термодинамическими параметрами пересольватации ионов Ni2+ и [NiEn]2+ (рис.1), которые мало различаются между собой.
Необходимость учета различий в сольватации отдельных фрагментов молекулы лиганда и степени их участия в комплексообразовании отмечается в работах [5,11] при рассмотрении применимости координационной модели сольватации для анализа влияния смешанного растворителя на комплексообразование. Чем больше размеры углеводородного фрагмента в молекуле лиганда, тем более значительно изменяется сольватация его молекулы при замене растворителя и тем больше молярная величина ïDtrHLï. Однако сольватация донорных групп определяется, главным образом, природой донорных атомов и сравнительно мало зависит от строения углеводородного фрагмента и его сольватации.
Таким образом, изменение теплового эффекта реакции комплексообразования, вызываемое заменой растворителя, можно рассматривать как эффект десольватации донорного атома лиганда и занимаемого им координационного места в сольватной оболочке иона металла. Фактор пересольватации донорного атома в большинстве случаев является превалирующим.
Рассмотрим в качестве примера термодинамику образования аминных комплексов Ni(II) в бинарных растворителях вода-ДМСО. Перенос аммиака из водного раствора в диметилсульфоксид эндотермичен, что свидетельствует о более слабой сольватации NH3 апротонным растворителем. Экзотермичность координации первой и второй молекулы аммиака растет с ростом содержания ДМСО в растворе: при XДМСО = 0,9 мол. доли прирост теплового эффекта комплексообразования составляет 13,1 ¸ 14,0 кДж/моль, что примерно соответствует по абсолютной величине энтальпии пересольватации NH3 в этой системе ΔtrHoH2O®ДМСО = 12,4 кДж/моль. Таким образом, весь прирост теплового эффекта реакции обеспечивается, по-видимому, более слабой сольватацией аммиачного лиганда в среде неводного растворителя.
При образовании комплексов с этилендиамином, который координирован по двум аминогруппам, прирост теплового эффекта реакции комплексообразования более значителен и составляет δ = 21 кДж/моль, т.е. в 1,5...1,6 раз больше, чем в случае образования моно-аммиакатов. Молекула этилендиамина, судя по значениям ΔtrHo, в ДМСО десольватирована в большей степени, чем молекула аммиака. Отметим, что энтальпии координации первой и второй молекул обоих лигандов практически одинаковы по величине в каждом из растворителей.
Рассмотренный пример показывает возможность прогнозирования энтальпий комплексообразования в неводных растворителях на основе сопоставления термодинамических характеристик реакций, в которых участвуют лиганды с одинаковыми донорными группами. Метод аддитивно-групповых вкладов указывает на закономерную связь термодинамических характеристик реакции с функциями сольватации лиганда и строением координационного соединения. Сравнительный анализ термодинамических характеристик образования различных комплексов в водно-органических средах позволяет также делать выводы о наличии в комплексах не координированных донорных групп [12].
Таблица.
Параметры ступенчатых реакций образования аминных комплексов Ni(II) в бинарных растворителях вода-ДМСО и энтальпии переноса реагентов (TPTB приближение) кДж/моль, Т=298К.
XДМСО мол.д. | -DrHo [NiEn] ±1 [13] | -DrH2o [Ni(En)2] ±1 | -DrHo [Ni(NH3)] ±0.4 [13] | -DrH2o [Ni(NH3)2] ±1 | DtrHo (En) ±0.3 H2O®ДМСО [14] | DtrHo(NH3) H2O®ДМСО [15] | DtrHo(Ni2+) H2O®ДМСО | |
[16, 3] | [16, 17] | |||||||
0 | 36.7 | 39.2 | 15.9 | 15.6 | 0 | 0 | 0 | 0 |
0.05 | 38.5 | 39.2 | 16.9 | 18.2 | - | 1.0 | 3.2 | 3.6 |
0.1 | 39.7 | 40.4 | 18.4 | 18.4 | 3.2 | 1.8 | 3.7 | 0.0 |
0.2 | 43.0 | 43.3 | - | - | 8.3 | - | -13.4 | -12.7 |
0.3 | 46.0 | 46.4 | 21.7 | 23.0 | 13.2 | 5.9 | -31.9 | -31.0 |
0.4 | 47.5 | 49.2 | 22.8 | 23.7 | 17.7 | 7.5 | -46.7 | -45.4 |
0.5 | 49.5 | 51.6 | 24.3 | 25.0 | 20.9 | 8.6 | -59.1 | -57.7 |
0.7 | 54.0 | 54.4 | 27.0 | 27.8 | 25.9 | 11.2 | -63.3 | -63.0 |
0.9 | 58.0 | 60.3 | 29.0 | 29.6 | 30.2 | 12.4 | -61.8 | -62.2 |
0.983 | - | - | - | - | 32.0 | 12.5 | - | - |
1.0 | - | - | - | - | 32.5 | 12.6 | -63.5 | -62.9 |
прирост d | -21.3 | -21.1 | -13.1 | -14.0 |
|
|
|
|
Литература
1. В кн. "Достижения и проблемы теории сольватации. Структурно-термодинамические аспекты". Сер. "Проблемы химии растворов" / Под ред. М: Наука. 1998. -247с. / Гл.6. С.172-205.
2. // Журн. общей химии. 1999. Т.69. Вып. 9. С.1421.
3. Нищенков . ... канд. хим. наук. 02.00.01. Иваново. 1986. ИХТИ. 277с.
4. , , // Журн. физич. химии. 1996. Т.70. Вып.7. С.1320.
5. , // Проблемы химии растворов и технологии жидкофазных материалов. Сб. науч. тр. Иваново. ИХР РАН 2001, С.119.
6. Lilley T.H. // Pure & Appl. Chem., 1994. V.66. N.3. P.429.
7. , , // Журн. общей химии, 1998. Т. 68. Вып. 5. С.763.
8. Пальчевский растворы электролитов. Л.: Из-во ЛГУ. 1984. 173с.
9. Р., , А// Коорд. химия. 2001. Т. 27, Вып.3. С.222.
10. Измайлов растворов. М.: Химия. 1976. с.179.
11. , // Журн. физич. химии. 1999. Т.73. Вып.3. С.465.
12. , «Термодинамика образования комплексов Ni(II)…» Журн. физич. химии . В печати.
13. , , // Коорд. химия. 1991. Т. 17, Вып.4. С.496.
14. , , // Журн. физич. химии. 1989. Т.63. Вып.1. С.244.
15. , , // Журн. физич. химии. 1988. Т.62. Вып.9. С.2568.
16. , , // Журн. физич. химии. 1984. Т.58. Вып.10. С.2475.
17. Hefter G., Marcus Y., Waghorn W.E // Chem.Rev. 2002. V.102. N8. P.2773.
