
Партнерка на США и Канаду по недвижимости, выплаты в крипто
- 30% recurring commission
- Выплаты в USDT
- Вывод каждую неделю
- Комиссия до 5 лет за каждого referral
Лекция 15
Кинетика фазовых переходов
Термодинамика позволяет установить температуру (и давление), при которой фазовое превращение происходит в равновесных условиях, но она не дает никакой информации о скорости протекания этого процесса, а это задача химической кинетики. Скорости фазовых превращений варьируются в широких пределах. С одной стороны, существуют превращения, протекающие с огромными скоростями и без гистерезиса в прямом и обратном направлениях (температура перехода при охлаждении и нагревании образцов совпадает). Другим крайним случаем являются переходы, скорость протекания которых можно измерить лишь в геологической шкале времени. Например, обсидиан ‑ стеклообразный минерал ‑ должен превращаться в одну из кристаллических модификаций SiO2, но из того факта, что такой минерал существует в природе, следует, что этот фазовый переход происходит крайне медленно. Большинство фазовых переходов занимают промежуточное положение между этими двумя крайними случаями, и их протекание связано с возникновением некоторого гистерезиса. Иначе говоря, высокотемпературная фаза может быть в различной степени переохлаждена до начала фазового превращения в низкотемпературную модификацию.
Скорость фазовых переходов варьируется в широких пределах и определяется целым рядом факторов. На рисунке 1 схематически изображена температурная зависимость скорости превращения низкотемпературной полиморфной модификации I в высокотемпературную II и обратного превращения. Вблизи равновесной температуры фазового перехода Тс скорости протекания как прямого, так и обратного превращения малы (область А). Поскольку в этой области ∆G ≈ 0, то движущая сила фазовых превращений в обоих направлениях мала. При температурах, удаленных от Тс, скорости превращений возрастают (области В, С). Максимальная скорость превращения II → I достигается при некоторой степени переохлаждения (при температуре Тм). Если бы было возможно переохладить высокотемпературную фазу II ниже Тм скорость превращения понизилась бы (область D). Часто максимум скорости превращения экспериментально обнаружить не удается, т. е. скорость превращения продолжает постоянно увеличиваться с понижением температуры (область С), хотя теоретически при более низких температурах точка Тм все же должна существовать. Выше Тс на кривой зависимости скорости превращения I → II соответствующий максимум отсутствует. Скорость этого процесса постоянно возрастает с температурой (область В).
|
Рисунок 1 – Температурная зависимость скоростей фазовых переходов первого рода из низкотемпературной модификации I в высокотемпературную II и в обратном направлении |

Общий вид зависимостей, приведенных на рисунке 1, и наличие максимума при Тм можно объяснить, если учесть следующие два фактора: 1) влияние температуры на скорость реакции в соответствии с уравнением Аррениуса; 2) зависимость разности свободных энергий фаз I и II от температуры. Совместное действие этих двух факторов дает основание сделать следующие выводы:
1) Уравнение Аррениуса применительно к кинетике химических реакций имеет вид
u = A exp(-E/RT) (1)
где Е — энергия активации превращения. Согласно этому уравнению, скорость реакции должна быстро возрастать с повышением температуры, что и происходит в областях D и В (рисунок 1). Для анализа уравнения (1) прологарифмируем его:
lg (u) = lg А - (E/RT) lg e (2)
и построим график зависимости lg (u) от 1/Т. Если скорость процесса подчиняется уравнению Аррениуса, то в этих координатах получаем прямую с наклоном (-E/R)lge, которая отсекает на оси ординат отрезок lgA.
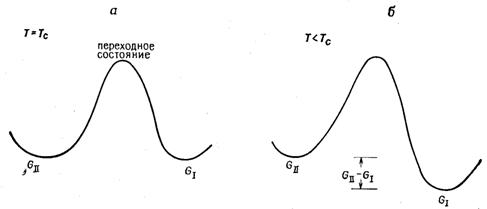
2) Разность свободных энергий двух полиморфных модификаций I и II ∆GI-II (так называемый термодинамический стимул) является движущей силой превращения (рисунок 2). В точке Тс GI = GII, и, следовательно, при этой температуре движущая сила как прямого, так и обратного перехода равна нулю (рисунок 2 а). При любой другой температуре GI ¹ GII, и превращение протекает преимущественно в одном из направлений (рисунок 2 б). В некотором гипотетическом идеальном случае Н и S обеих фаз не зависят от температуры), и ∆GI-II при любой температуре ‑ линейная функция разности температур Тс-Т:
![]() (3)
(3)
![]() (4)
(4)
или
![]() (5)
(5)
|
Рисунок 2 – Разность свободных энергий полиморфных фаз I и II |
Комбинация приведенных выше факторов определяет скорость протекания фазового превращения. Не делая количественных оценок, можно сказать, что второй фактор доминирует при температурах, близких к Тс, где величина термодинамического стимула превращения мала. При температурах, заметно отличающихся от Тс, более важное значение приобретает первый фактор, особенно в областях В и D (рисунок 1), где скорость превращений экспоненциально возрастает с температурой.
Основные трудности возникают при желании количественно описать влияние этих двух факторов (особенно второго). Традиционный подход заключается в соотнесении термодинамического стимула ∆GI-II (второй фактор) со скоростью образования зародышей продукта фазового превращения. Большинство фазовых переходов первого рода или реконструктивного типа идут по двухстадийному механизму (стадия зародышеобразования + стадия роста кристаллов), причем лимитирующим процессом является начальный процесс образования зародышей продукта превращения. Хотя теория процесса зародышеобразования развита весьма хорошо, трудно, а может быть, и невозможно применить ее для количественных расчетов, поскольку значения некоторых параметров (например, поверхностная энергия зародышей) неизвестны. Рассмотрим кратко основные положения этой теории.
Критический размер зародышей
Начнем рассмотрение с фазы, которая находится в состоянии термодинамического равновесия. Фазовое превращение, т. е. реакция образования другой полиморфной модификации, не может протекать в заметной степени в таких условиях, поскольку подобный процесс неизбежно связан с общим увеличением свободной энергии (например, переход I → II на рисунке 2 б).
Предположим, что внешние параметры системы (Р и Т) изменились и исходная полиморфная модификация больше не находится в равновесном состоянии. Согласно законам термодинамики, в этих условиях должно начаться превращение в полиморфную модификацию с более низкой свободной энергией. Однако если такое превращение протекает по упомянутому выше двухстадийному механизму, то его скорость может оказаться весьма низкой. Начальным процессом образования новой фазы является возникновение микрозародышей либо на поверхности, либо во всем объеме исходной полиморфной фазы. Протеканию процесса зародышеобразования препятствует то обстоятельство, что поверхностная энергия зародышей вносит дополнительный положительный вклад в величину свободной энергии системы. Поэтому суммарное изменение свободной энергии ∆Gn является комбинацией уменьшения свободной энергии за счет образования зародышей продукта превращения и увеличения свободной энергии за счет вклада поверхностной энергии зародышей. Если исходная фаза и продукт превращения имеют различные мольные объемы, то в выражение для свободной энергии входит дополнительный член, учитывающий энергию напряжения. Для простоты положим, что ∆V = 0 и энергия напряжений равна нулю. Пусть ∆GV ‑ изменение объемной свободной энергии, приходящееся на единицу объема зародыша по отношению к исходной фазе, a ∆Ga ‑ поверхностная свободная энергия, приходящаяся на единицу поверхности зародыша. Для сферических зародышей радиуса r имеем
![]() (6)
(6)
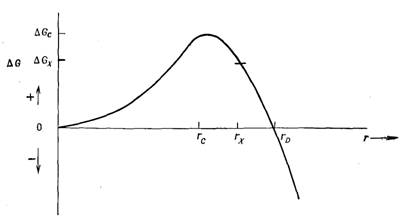
Для малых r ∆Gn положительна, так как 4nr2∆Ga > 4/3pr3∆GV (рисунок 3). Однако с ростом r ∆Gn проходит через максимум (∆GC) при r = rс и уменьшается до нуля при r = rD. Для r >rD ∆Gn отрицательна. Это дает основание считать, что для того, чтобы зародыши были устойчивыми, их размер должен быть выше некоторого критического размера. На первый взгляд кажется, что в качестве критического размера может быть выбран радиус rD (при r > rD ∆Gn становится отрицательным). По кинетическим соображениям, однако, критический радиус соответствует величине rс, так как при r > rc ∆Gn n начинает уменьшаться с ростом r. Чтобы объяснить это положение, рассмотрим поведение зародыша радиуса rx (rc < rx < rD), обладающего свободной энергией +GX. Хотя такой зародыш термодинамически неустойчив, он устойчив кинетически, так как, если бы зародыш начал растворяться и его радиус r уменьшался бы, это привело бы к возрастанию значения ∆Gn в сторону ∆GC. Поэтому если зародыши радиуса r > rс образовались, то они кинетически устойчивы и, следовательно, будут расти. Фактически зародыши радиуса rх метастабильны, так как для их растворения необходимо преодолеть активационный барьер, равный ∆GC ‑ ∆GX. Зародыши радиуса r < rc нестабильны, поскольку для их растворения не требуется преодолевать энергетических барьеров. Зародыши радиуса r > rD термодинамически стабильны.
|
Рисунок 3 – Изменение свободной энергии зародышей в зависимости от их радиуса |
Значение rс (рисунок 3) можно найти из условия d∆G/dr = 0,
т. е.
![]()
![]() (7)
(7)
Критическую величину избыточной свободной энергии получим после подстановки выражения для rс в уравнение (6):
![]() (8)
(8)
Теперь становится ясным, почему зародышеобразование затруднено вблизи температуры Тс: при Т → Тс ∆GV → 0 и, согласно уравнениям (7) и (8), rс и ∆GC принимают очень большие значения. Зависимость изменения rс от температуры можно получить путем подстановки (5) в уравнение (7):
![]() (9)
(9)
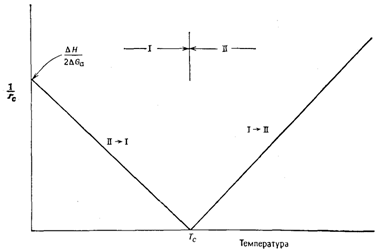
График зависимости rC-1 от Т (рисунок 4) представляет собой прямую, пересекающую ось температур в точке Тс, когда rс → ¥. Две ветви графика r-1(Т) отвечают двум направлениям протекания фазовых превращений I → II.
|
Рисунок 4 – Зависимость критического размера зародышей от температуры |
Скорость зародышеобразования
Зародыши продукта фазового превращения образуются благодаря тепловому движению атомов, поэтому скорость зародышеобразования ‑ функция температуры
![]() (10)
(10)
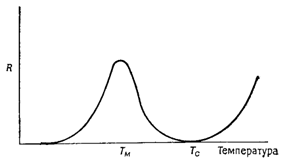
Суммарная энергия активации процесса ∆Gc + ∆Ga учитывает вклад критической свободной энергии ∆GC, которую необходимо превысить для образования устойчивых зародышей, и энергии активации перескока отдельных атомов ∆Ga, участвующих в зародышеобразовании. Поскольку ∆GC зависит от температуры [уравнения (8) и (5)], скорость зародышеобразовавания R изменяется с температурой таким образом, как показано на рисунке 5. При фазовом переходе в направлении II → I скорость зародышеобразования R проходит через максимум при некоторой температуре Тм (ниже Тс) и стремится к нулю как при Т → 0 К, так и при Т → ТС. При фазовом переходе I → II R быстро растет с температурой (при Т > ТС).
|
Рисунок 5 – Влияние температуры на скорость зародышеобразования R |
Вышесказанное вполне подтверждает, что уравнение (10) и рисунок 5 во многих случаях качественно верно описывают кинетику зародышеобразования. Однако скорость зародышеобразования трудно измерить экспериментально и количественно проверить обоснованность теоретических выводов. Трудности частично связаны с тем, что на скорость зародышеобразования заметно влияет наличие примесей, дислокаций, поверхностей раздела фаз и т. п. Следует различать два типа зародышеобразования. Под гомогенным зародышеобразованием понимают процесс, протекающий в объеме однородной исходной фазы; это случайный процесс, зависящий от термических флуктуации состава исходной фазы. Гораздо легче протекает гетерогенное зародышеобразование, при котором зарождение новой фазы идет в основном на дефектах и позициях с высокой локальной свободной энергией.
Общая скорость превращения. Уравнение Аврами
Экспериментально намного более просто провести измерение общей скорости фазового превращения, чем выделять отдельные стадии этого процесса (образование зародышей и их рост), особенно если исследуемое вещество ‑ порошок. Для описания экспериментальных данных часто пользуются уравнением Аврами (его называют также уравнением Аврами ‑ Джонсона ‑ Мела ‑ Ерофеева):
![]() (11)
(11)
Здесь α ‑ объемная доля продукта превращения, k ‑ константа скорости процесса (кинетическая константа), п ‑ постоянная, зависящая от природы процессов зародышеобразования и роста кристаллов; п и k можно определить, дважды прологарифмировав уравнение (11):
![]() (12)
(12)
и построив график зависимости lglg(l/l—а) от lg t.
Применение уравнения Аврами ‑ удобный способ обработки экспериментальных данных: для этого не требуется делать никаких предположений, а при благоприятных обстоятельствах (например, если получено целочисленное значение п) можно сделать вывод о механизме превращения. Так, если при полиморфном превращении п = 3, можно показать, что зародыши новой фазы начинают образовываться лишь в ходе фазового перехода. Если п = 4, то это указывает на существование зародышей новой фазы уже в матрице исходного вещества. Даже в тех случаях, когда полученное значение п не дает возможности сделать такие качественные выводы, уравнение Аврами тем не менее позволяет определить такой важный параметр, как константа скорости реакции k. Если измерения скорости фазового превращения проведены в некотором интервале температур, то с помощью аррениусовской зависимости lgk от 1/Т можно определить энергию активации процесса.
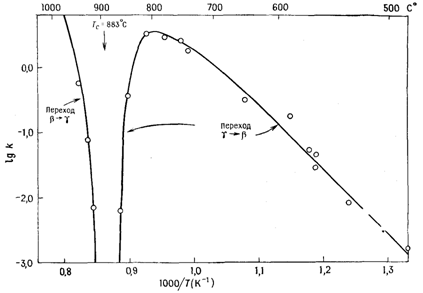
В качестве примера рассмотрим изображенную на рисунке 6 зависимость lgk от 1/T для фазового перехода b«g-Li2ZnSiО4. Кинетика этого превращения была изучена в широком интервале температур (480-940 ºС) как для прямого, так и обратного превращения. При температурах намного ниже Тс (883 °С) экспериментальные данные ложатся на прямую, что приводит к значению энергии активации этого превращения, равному 185 кДж/моль. В интервале температур 780—950 °С скорость превращения понижается, особенно резко вблизи Тс. Такой ход зависимости подчеркивает важную роль термодинамических факторов, т. е. разности свободных энергий обеих полиморфных модификаций, для кинетики фазовых переходов.
|
Рисунок 6 – График аррениусовской зависимости скорости фазового перехода b«g-Li2ZnSiО4 |
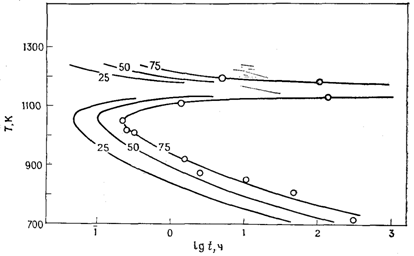
Диаграммы время – температура ‑ превращение (ТТТ-диаграммы)
Экспериментальные исследования кинетики фазовых переходов часто нацелены лишь на определение скоростей процессов в зависимости от температуры без изучения деталей механизма превращения. При этом вовсе не ставится цель использовать корректные кинетические уравнения. В таких случаях кинетические данные удобно представлять в виде ТТТ-диаграмм, на которых откладывают время (в логарифмических координатах), необходимое для заданной степени превращения (например, 25 %) в зависимости от температуры. В качестве примера на рисунке 7 приведена ТТТ-диаграмма для превращения b«g-Li2ZnSiО4 (сравните с рисунком 6). На ТТТ-диаграмме нанесены три кривые, отвечающие трем разным степеням превращения: 25, 50 и 75 %. Видно, что скорости превращения в направлении g → b становятся максимальными при температуре ~ 1060 К (т. е. при ~ 790 °С), а при ~ 1150 К скорости превращения в обоих направлениях крайне малы. Быстрое увеличение скорости b → g-перехода начинается выше ~ 1180 К.
|
Рисунок 7 – ТТТ-диаграммы для фазового перехода b«g-Li2ZnSiО4 |
TTТ-диаграммы, аналогичные приведенным на рисунке 7, применяются при исследованиях многих металлов и сплавов. Они также имеют важное значение при изучении фазовых превращений с участием неорганических веществ.
Факторы, влияющие на кинетику фазовых переходов
Во многих книгах по кинетике основное внимание уделено реакциям в газообразном и жидком состояниях; о твердофазных реакциях, как правило, упоминается весьма редко, в основном в связи с применением твердых тел в качестве катализаторов реакций с участием жидких и (или) газообразных веществ. Причина такого невнимания заключается не в том, что твердофазные реакции имеют небольшое практическое значение; такой вывод был бы совершенно неверным. Причина в другом: механизм их протекания чрезвычайно сложен. Строгая количественная интерпретация результатов твердофазных реакций весьма трудна, если вообще возможна. Например, концепция о порядке реакции ‑ центральный момент кинетической теории реакций газообразных веществ ‑ к большинству твердофазных превращений не применима.
Факторы, влияющие на кинетику обоих типов превращений, во многом аналогичны. Фазовые переходы проще твердофазных реакций по двум причинам:
1) фазовый переход обычно не сопровождается изменением состава фаз. В твердофазных же реакциях изменение состава фаз происходит неизбежно, поэтому при исследовании последних важным фактором является учет диффузии ионов в твердом теле;
2) жидкая и газовая фазы, как правило, не оказывают заметного влияния на фазовые переходы в твердом состоянии.
Исключение составляют случаи, когда они катализируют фазовые превращения в твердом веществе. Однако в реакциях между твердыми телами роль газовой фазы иногда очень существенна из-за того, что в ходе реакций газы могут поглощаться или, наоборот, выделяться. Другими словами, газовая фаза – это среда для переноса вещества от одной частицы к другой.
Сравнение кинетики и механизма фазовых переходов, с одной стороны, и реакций с участием жидкой и газовой фаз ‑ с другой, показывает, что между ними существуют принципиальные различия. Наиболее существенные из них связаны с размером объектов, участвующих в этих процессах. Реакции в жидкостях и газах идут с участием небольшого числа атомов в каждом элементарном акте. Например, реакция атома водорода и атома хлора приводит к образованию молекулы хлороводорода:
Н + .С1 → НС1
В большинстве фазовых переходов в формировании устойчивых зародышей продукта превращения участвует, как правило, гораздо большее число атомов. Например, при фазовом переходе анатаза ТiO2 в рутил зародыши рутила образуются как на поверхности, так и в объеме кристалла анатаза, причем размер зародышей увеличивается со временем. Предположим, что при некоторых условиях самый мелкий зародыш рутила, который может независимо существовать, имеет диаметр 50 Å; даже такой зародыш содержит около 5000 атомов.
Другое различие между реакциями в газовой и твердой фазах состоит в том, что на последние заметное влияние оказывают поверхностные явления: зародыши новой фазы часто возникают на поверхности исходного кристалла. Кинетика фазовых превращений зависит от общей поверхности кристаллов исходной фазы, а следовательно, от того, идет ли процесс на монокристалле или в порошкообразном материале. Как было отмечено выше, поверхностные явления определяют устойчивость зародышей образующегося продукта. Поверхностная энергия зародышей вносит положительный вклад в свободную энергию системы. Если этот вклад превышает выигрыш свободной энергии в объеме зародыша, то такой зародыш будет неустойчивым, и он начнет растворяться. В отличие от этого поверхностные явления практически не влияют на кинетику реакций, происходящих в газовой фазе и в жидкостях, за исключением случаев, когда твердые вещества выступают в роли катализаторов.
На кинетику фазовых переходов оказывают влияние многие другие факторы:
1. природа образца (монокристалл или порошок) и площадь поверхности;
2. температура кинетического исследования по сравнению с равновесной температурой превращения;
3. энергия активации и прочность связей, которые необходимо разрушить;
4. предэкспотенциальный множитель А;
5. изменение объема ∆V;
6. давление Р;
7. механизм фазового перехода.
Уже отмечалось значение величины поверхности образца; существенной оказывается и температура, при которой проводится кинетическое исследование, особенно в сравнении с равновесной температурой превращения. На рисунке 6 показано влияние величины энергии активации Е и предэкспоненциального множителя А на кинетику фазового перехода. Высокое значение А (при T-1 → 0 lgA = lgk) приводит к быстрому протеканию превращения при высоких температурах. Если же при этом энергия активации Е невелика, то высокая скорость характерна для такого превращения и при низких температурах. Энергия активации может служить для оценки прочности химических связей, которые необходимо разрушить, чтобы произошло превращение. Поэтому большинство реконструктивных фазовых переходов характеризуются высокими значениями Е и протекают медленно. Энергия активации деформационных фазовых превращений, как правило, невелика, и они протекают довольно быстро.
Кинетика фазовых переходов зависит также от разности объемов ∆V обеих полиморфных модификаций. Из теории абсолютных скоростей следует
![]() (13)
(13)
где ∆V — разность объемов исходной фазы и промежуточного продукта. Очевидно, что скорость превращения увеличивается с уменьшением ∆V. Многие фазовые переходы термодинамически осуществимы лишь при высоких давлениях. Однако, как следует из уравнения (13), скорость таких переходов с ростом давления уменьшается. В качестве примера можно упомянуть синтез алмаза из графита. Алмаз термодинамически стабилен лишь при высоких давлениях. Поэтому повышенное давление, с одной стороны, необходимо для получения алмаза, а с другой ‑ оно приводит к замедлению скорости образования алмаза.
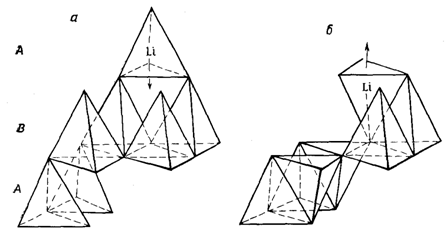
К важным факторам, определяющим скорость фазового перехода, относится механизм превращения. В качестве примера можно сравнить реконструктивный и деформационный механизмы превращений. Лимитирующей стадией многих фазовых переходов является процесс зародышеобразования. Этот процесс зависит от многих факторов, например от природы твердых тел (монокристаллические и порошкообразные вещества), наличия и природы дефектов кристаллической решетки (вакансии, дислокации, примеси и т. п.), состава атмосферы. На скорость образования зародышей новой фазы заметно влияет также различие кристаллической структуры обеих полиморфных модификаций. Если кристаллические решетки обеих фаз незначительно отличаются друг от друга и фазовый переход состоит в том, что претерпевает изменение в основном второе координационное окружение, то процесс зародышеобразования протекает легко. Если же в ходе превращения идет существенная перестройка кристаллической решетки, то зародышеобразование протекает медленно. Промежуточное положение занимают так называемые топотактические превращения, при которых хотя и сохраняется определенная ориентация кристаллов обеих полиморфных модификаций, тем не менее, для протекания превращения требуется существенная перестройка кристаллической решетки. Примерами таких фазовых переходов могут служить превращения, при которых анионная подрешетка кристалла остается неизменной, а катионная подрешетка существенно перестраивается; при этом зародышеобразование не слишком затруднено. Фазовый переход топотактического типа был обнаружен в ходе превращения b«g-Li2ZnSiО4 (рисунок 8). b-Li2ZnSiО4 имеет вюртцитоподобную кристаллическую структуру, в которой катионы упорядоченно размещаются в тетраэдрических пустотах гексагональной плотнейшей упаковки ионов кислорода. При фазовом переходе в g-Li2ZnSiО4 кислородные слои остаются без изменения (лишь слегка изгибаются), а половина катионов перемещается в незанятые тетраэдрические позиции. Это приводит к переориентации некоторых тетраэдров МО4.
Необходимо обратить внимание на соблюдение принципа обратимости микросостояний. Такой подход оказался весьма плодотворным при изучении кинетики газофазных реакций. Видимо, он полезен и при изучении кинетики твердофазных превращений. Согласно принципу обратимости микросостояний, кинетика прямой и обратной реакций характеризуется константами k1 и k-1:
|
|
Рисунок 8 – Топотактический механизм фазового перехода b«g-Li2ZnSiО4 |
Для реакций в газовой фазе можно сформулировать два следствия из данного принципа. Во-первых, константа скорости превращения равна разности между k1 и k-1. Во-вторых, это превращение не идет до конца ни в одном из направлений. В определенный момент система достигает равновесия, т. е. скорости прямой и обратной реакций становятся равными. В случае применения принципа обратимости микросостояний к твердофазным превращениям возникают некоторые отличия от рассмотренного выше подхода к реакциям в газовой фазе. Первое следствие по-прежнему выполняется: скорость твердофазного превращения равна разности скоростей прямой и обратной реакций. А вот второе следствие к твердофазным превращениям не применимо. В условиях термодинамического равновесия твердофазные реакции и фазовые превращения в твердом состоянии должны протекать необратимо в одном из направлений. В противном случае будет нарушено правило фаз. Чтобы показать это, обратимся к уже упоминавшемуся выше фазовому превращению b«g в Li2ZnSiО4. Поскольку Li2ZnSiО4 ‑ конгруэнтно плавящаяся термодинамически стабильная фаза, это вещество можно рассматривать как однокомпонентную систему (С=1). Без учета равновесия с газовой фазой и при нормальном давлении правило фаз для конденсированной системы можно записать (Р = 2) могут находиться в равновесии лишь при строго фиксированной температуре Тс (Р = 0). При любой другой температуре термодинамически стабильной оказывается только одна из полиморфных модификаций (b или g). Такие рассуждения должны привести нас к однозначному выводу о том, что для твердофазных реакций и фазовых превращений принцип обратимости микросостояний может быть применим лишь частично: согласно правилу фаз Гиббса, твердофазные реакции должны идти до конца в одном из направлений.
