

Партнерка на США и Канаду по недвижимости, выплаты в крипто
- 30% recurring commission
- Выплаты в USDT
- Вывод каждую неделю
- Комиссия до 5 лет за каждого referral
Лекция № 15
ХРОМАТОГРАФИЧЕСКИЕ МЕТОДЫ АНАЛИЗА
1.Классификация хроматографических методов анализа
Хроматография – это не только метод разделения, основанный на распределении веществ между подвижной и неподвижной фазами, но и метод качественного и количественного анализа.
В качестве подвижной фазы используют газ или легколетучую жидкость. Неподвижная фаза – это твердое пористое вещество или вязкая нелетучая жидкость. Неподвижная фаза наносится на стеклянную или полимерную пластинку, или помещается в тонкую стеклянную или металлическую трубку. Смесь, которую надо разделить, вместе с подвижной фазой, перемещается через неподвижную фазу. Те компоненты, которые меньше взаимодействуют с неподвижной фазой, перемещаются быстрее. Если компоненты хорошо взаимодействуют с неподвижной фазой, они перемещаются медленно.
По агрегатному состоянию подвижной и неподвижной фаз различают газовую и жидкостную хроматографии. По механизму взаимодействия компонентов с неподвижной фазой хроматографические методы подразделяют на распределительную, ионообменную, адсорбционную и др. По технике выполнения – на колоночную и плоскостную (бумажную и тонкослойную).
Графическое изображение распределения веществ в подвижной фазе на выходе из хроматографической колонки или на плоскости после прохождения подвижной фазы через всю пластинку, называют хроматограммой.
По способу получения различают элюентную, вытеснительную и фронтальную хроматографии.
Элюентная хроматография происходит от слова «элюент» - это подвижная фаза, которая переносит анализируемую смесь через неподвижную фазу. Элюат – это подвижная фаза, содержащая разделенные компоненты на выходе их колонки.
Элюент (растворитель) должен как можно меньше сорбироваться на поверхности неподвижной фазы. Пробу вводят в колонку или наносят на пластинку вместе с растворителем и дальше продолжают пропускать через колонку чистый растворитель. Компоненты анализируемой смеси перемещаются вдоль колонки (пластины) с разными скоростями в соответствии с их сорбируемостью. На выходе из колонки сначала появляется наименее сорбируемый компонент, затем следующий и т. д. В этом случае хроматограмма имеет вид (рис. 1). Этот вид хроматографии наиболее распространен.
Вытеснительная хроматография. При выполнении вытеснительной хроматографии сначала в колонку вводят раствор с разделяемыми компонентами, затем через колонку пропускаю растворитель с большой сорбируемостью (вытеснитель). В результате первым по колонке перемещается компонент с минимальной сорбируемостью, затем – с большей и т. д. Последним из колонки выходит вытеснитель. Примером вытеснительной хроматографии может быть ионообменная хроматография. В этом случае каждый компонент выделяется в чистом виде, но не количественно, так как зоны компонентов на хроматограмме не разделены участками чистого сорбента (рис.2).




Рис.1. Хроматограмма элюентной хроматографии Рис.2. Хроматограмма вытеснительной
хроматографии
Фронтальная хроматография осуществляется следующим образом. В колонку непрерывно подают раствор разделяемых компонентов, сорбируемость которых увеличивается в ряду А < В < С. Из колонки сначала выходит чистый растворитель, затем наименее сорбируемый компонент А, затем смесь компонентов А и В и т. д. При этом способе проведения хроматографии в чистом виде (но не количественно) можно выделить только один, наименее сорбируемый компонент. Хроматограмма в этом случае имеет вид, показанный на рис.3.
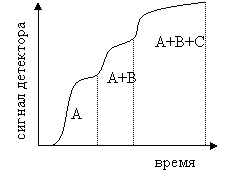

Рис.3 Хроматограмма фронтальной хроматографии
2. Хроматографические параметры
Классическая хроматограмма имеет вид (рис. 4).
Время от момента ввода пробы до регистрации максимума на хроматограмме называется временем удерживания. На рис. 4 это tR1 и tR2. Значение tm равно времени прохождения через колонку несорбируемого компонента.
Время удерживания не зависит от количества пробы, а зависит лишь от природы вещества и сорбента, от качества заполнения колонки, то есть на каждой конкретной колонке для одного и того же вещества время удерживания есть величина постоянная. Время удерживания является качественным аналитическим сигналом. Чтобы учесть особенности заполнения сорбентом каждой конкретной колонки вводят понятие «исправленное время удерживания»:
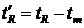 .
.
Количество вещества, вымываемое из колонки, можно найти по площади под кривой элюирования:
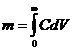 ,
,
где С – концентрация, моль/мл;
V – объем, мл.
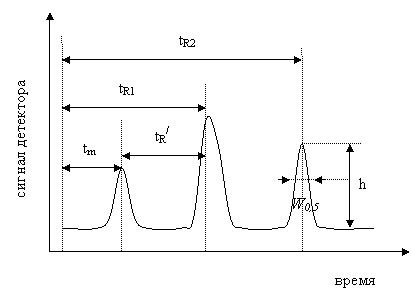

Рис.4 Классическая хроматограмма
Обычно хроматографический пик рассматривают как равнобедренный треугольник, площадь которого находят как произведение высоты пика h на ширину пика на половине его высоты W0,5: ![]() .
.
Количественные определения проводят по методу градуировочного графика или методы внешнего стандарта, но точность этих методов не очень высока, так как при вводе пробы в хроматографическую колонку часть ее теряется.
Более точный метод – метод внутреннего стандарта. В пробу вводят строго известное количество какого-либо вещества, которое не взаимодействует с определяемым компонентом и хорошо разделяется с ним в колонке. При этом желательно, чтобы время выхода определяемого вещества и внутреннего стандарта не очень сильно различались. Определяют площадь пика внутреннего стандарта и по пропорции рассчитывают количество определяемого вещества:
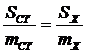 .
.
Метод будет точен тогда, когда чувствительность детектора к определяемому веществу и к внутреннему стандарту одинаковая.
Метод нормировки используют тогда, когда детектор одинаково чувствителен ко всем компонентам пробы и на хроматограмме регистрируются пики всех компонентов пробы. В этом случае определяют площадь каждого пика и рассчитывают их сумму. Процентное отношение площади пика к общей площади всех пиков в этом случае дает массовую долю компонента.
Если чувствительность детектора к компонентам различная, но все пики регистрируются на хроматограмме, при расчетах используют поправочные коэффициенты.
3. Плоскостная хроматография
К плоскостным видам хроматографии относят бумажную и тонкослойную хроматографии. Эти два вида просты по технике исполнения, не требуют сложных приборов.
На пластинку с сорбентом (носителем) наносят на линию старта индивидуальные вещества, которые предполагают обнаружить в пробе, и пробу. Пластинку погружают вертикально в хроматографическую камеру, на дно которой налит растворитель (подвижная фаза). При этом уровень растворителя не должен превышать линии старта, лучше, если он не доходит до нее на 0,5 – 1 см. Подвижная фаза поднимается под действием капиллярных сил и увлекает с собой компоненты. Подвижная фаза не должна сорбироваться на поверхности носителя. Быстрее всего вверх поднимается вместе с растворителем наименее сорбируемый на носителе компонент. Наиболее сорбируемый компонент движется медленнее всего. После того, как линия растворителя поднимется до уровня 1 – 2 см от верхнего края пластинки, пластинку вынимают из хроматографической камеры, высушивают, затем обрабатывают различными реагентами для проявления хроматограммы. В результате получаем подобную картинку (рис.5):
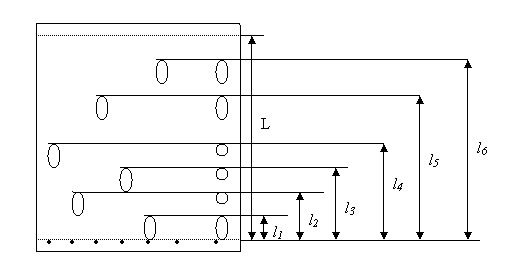

Рис.5 Тонкослойная хроматограмма
Относительная скорость перемещения компонентов 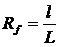 . Этот коэффициент зависит от природы компонента, носителя, растворителя, температуры, техники проведения эксперимента. В идеальном случае он не зависит от концентрации компонента и позволяет качественно идентифицировать компоненты.
. Этот коэффициент зависит от природы компонента, носителя, растворителя, температуры, техники проведения эксперимента. В идеальном случае он не зависит от концентрации компонента и позволяет качественно идентифицировать компоненты.
Количественный анализ проводят следующим образом: пятно на хроматограмме, содержащее определяемое вещество, вымывают из слоя сорбента, полученный раствор анализируют подходящим методом. Для количественного анализа необходимо точно знать объем наносимой пробы.
4. Газовая хроматография
Газовая хроматография – метод анализа, в котором разделение компонентов смеси основано на различии в равновесном распределении компонентов между газом – носителем (подвижная фаза) и твердой или жидкой неподвижной фазами. Она подразделяется на газо-жидкостную хроматографию, в которой вязкая нелетучая высококипящая жидкость наносится на твердый сорбент или на стенки тонкой трубки; и на газо-адсорбционную, в которой неподвижная фаза – это твердый сорбент, которым плотно заполнена хроматографическая колонка.
Анализируемый раствор в испарителе переводится в парообразное состояние и смешивается с потоком инертного газа-носителя (аргона или гелия). Эта смесь продвигается по колонке под действием непрерывно подаваемого газа – носителя. В колонке постоянно осуществляется сорбция и десорбция компонентов раствора, причем, чем сильнее сорбируется компонент на поверхности неподвижной фазы, тем медленнее происходит процесс десорбции, и тем дольше удерживается этот компонент в колонке. Чем сильнее различаются коэффициенты распределения компонентов, тем легче они разделяются.
Пары разделенных компонентов вместе с газом – носителем поступают в детектор хроматографа, в котором генерируется электрический сигнал, пропорциональный концентрации компонента.
Детекторы бывают двух видов: неселективные и селективные. К неселективным детекторам относятся детектор по теплопроводности (катарометр), детектор ионизации пламени (ДИП) и электрокондуктометрический детектор. Сигнал этих детекторов зависит не от природы компонента, а только от его количества.
К селективным детекторам относятся термоионные, электронозахватные, пламенно-фотометрические и т. п. Сигнал этих детекторов зависит как от природы компонента, так и от его количества.
В катарометре имеются две отделенные друг от друга вольфрамовые или платиновые нити, через которые пропускают электрический ток. Одна нить находится в потоке чистого газа – носителя, а другая – в потоке с анализируемым веществом. Если состав потоков отличается, то они в различной степени охлаждают нити. В зависимости от температуры меняется и сопротивление нити, возникает разность потенциалов, которая регистрируется прибором. Чувствительность катарометра зависит от природы газа – носителя. Для азота, аргона, углекислого газа – это 10-5 г, для водорода и гелия – около 10-6 – 10-7 г.
Детектор ионизации пламени более чувствителен, чем катарометр. Смесь разделенных после колонки компонентов попадает в пламя водородной горелки, находящееся между электродами. Вещество в пламени ионизируется, вследствие чего ток между электродами возрастает. Увеличение электропроводности пламени регистрируется прибором. Но ДИП чувствителен только к тем веществам, которые при температуре водородного пламени подвергаются ионизации.
Электрокондуктометрический детектор основан на том, что разделенная смесь после выхода из колонки попадает в зону печи, нагретой до 700 – 800 оС. Органические вещества сгораю, а продукты сгорания (углекислый, сернистый газ) поглощаются водой. Образуется раствор слабых кислот (угольной, сернистой), в результате чего электропроводность воды несколько повышается. Изменение электропроводности также регистрируется приборами.
Селективные детекторы могут быть еще более чувствительными. Так, термоионный детектор реагирует на соединения азота и фосфора в количестве 10-12 – 10-14 г. Чувствительности электронозахватывающего детектора для определения галогеносодержащих, металлоорганических, полиароматических соединений равна также 10-12 – 10-14 г.
Пламенно-фотометрический детектор основан на измерении интенсивности излучения продуктов атомизации разделенных компонентов в водородном пламени, его чувствительность 10-12 – 10-13 г.
Селективные детекторы применяют в том случае, если проводится серийный анализ одного или группы компонентов.
Если не удается определить вещества прямым детектированием, проводят непрямое (косвенное) детектирование. В этом случае элюент дает постоянный сигнал детектора, который ослабевает при прохождении через него компонентов анализируемой смеси. Таким образом, определяют щелочные металлы, не поглощающие в УФ-области, на фоне разбавленного раствора сульфата меди, имеющего высокое поглощение.
Возможно определение компонентов путем проведения послеколоночной реакции. Для повышения чувствительности и селективности определения в смесь разделенных веществ вводят спектрофотометрический реагент, который вступает с разделенными веществами в реакцию с образованием интенсивно окрашенных или флуоресцирующих соединений.
5. Жидкостная колоночная хроматография
Этот метод используется для разделения и определения тех веществ, которые разлагаются при нагревании. В этом случае подвижная фаза – жидкость, хроматография проводится при обычной температуре. Стеклянную колонку заполняют сорбентом, вводят пробу и промывают элюентом. Скорость классической жидкостной хроматографии очень мала. Большее распространение получила ВЭЖХ – высокоэффективная жидкостная хроматография, в которой элюент подается под давлением, в результате скорость прохождения пробы через колонку возрастает. К жидкостной хроматографии относятся адсорбционная, распределительная, ионообменная, эксклюзионная и другие виды хроматографии.
Адсорбционная жидкостная хроматография делится на нормально-фазовую (НФХ) и обращено-фазовую (ОФХ). В НФХ используют полярный сорбент и неполярную подвижную фазу. Неподвижная фаза за счет сил адсорбции удерживает вещества. Очень важен выбор подвижной фазы. Она должна обеспечивать эффективное разделение за приемлемое время. В ОФХ, наоборот, неполярный сорбент и полярная подвижная фаза.
Распределительная жидкостная хроматография основана на распределении вещества между двумя жидкостями. В колонке происходит многократная ступенчатая экстракция. Жидкую неподвижную фазу наносят на пористый инертный носитель и заполняют им колонку. При пропускании подвижной фазы с пробой, компоненты разделяются за счет различной растворимости в жидкой фазе. В подвижной фазе компоненты должны растворяться лучше, чем в неподвижной, иначе время удерживания будет очень большое. При этом часто в подвижную фазу добавляют третий компонент, снижающий различие в полярности фаз. Именно в этом случае удается достичь приемлемого разделения.
В НФХ неподвижная полярная фаза – вода, спирт, а неполярная подвижная фаза – гексан, изооктан, бензол. В ОФХ – наоборот.
Ионообменная хроматография основана на процессе замещения ионов, связанных с неподвижной фазой, ионами элюента, поступающими в колонку. Удается разделить ионы с зарядом одного и того же знака (катионы от анионов).
Эксклюзионная хроматография основана на разделении компонентов в соответствии с их размерами: в процессе разделения небольшие молекулы попадают в сетку полимера, в порах которого растворитель (неподвижная фаза) удерживает эти молекулы. Большие молекулы не могут проникнуть в полимерную сетку и вымываются вместе с подвижной фазой. Такую хроматографию еще называют молекулярно-ситовой.
