

Партнерка на США и Канаду по недвижимости, выплаты в крипто
- 30% recurring commission
- Выплаты в USDT
- Вывод каждую неделю
- Комиссия до 5 лет за каждого referral
Модуль 3. Лекция 10.
Применение других оптических методов в количественном анализе
Рефрактометрия. Сущность метода рассмотрена в книге 1 (гл. 20, раздел 20.2.5). Применение метода в количественном анализе основано на использовании зависимости между показателем преломления (светопреломления) n анализируемого раствора и содержанием X определяемого вещества в этом растворе. Для растворов различных веществ в широком диапазоне изменения концентраций зависимость показателя преломления от концентрации раствора может быть как линейной, так и нелинейной (рис. 8.23). Если в некотором интервале изменений X и n эта зависимость линейна, то выполняется соотношение (8.15):
X= (n-n0) /F, (8.15)
Где n – показатель преломления раствора, n0 – показатель преломления чистого растворителя (для воды nD20=1,3330; nD25=1,3325), F – фактор, равный величине прироста показателя преломления раствора при увеличении содержания растворенного вещества на 1%.
Фактор F находят экспериментально. Для этого измеряют значения показателей преломления n1 и n2 двух растворов с содержанием данного определяемого вещества соответственно X1 и X2, выраженного в процентах, после чего рассчитывают значение F по формуле (8.16):
F= (n2-n1) / (X2-X1). (8.16)
Значения n1 и n2 выбирают так, чтобы величина n испытуемого раствора лежала как можно ближе к n1 и n2, а сам интервал n2-n1 был бы по возможности минимальным.
Так, например, показатели преломления nD20 водного раствора хлорида кальция при его содержании X1=2,40% и X2=5,00% равны соответственно n1=1,3360 и n2=1,3390. Тогда получаем:
F= (n2-n1) / (X2-X1) = (1,3390-1,3360) / (5,00-2,40) = 0,001154.
Поскольку показатель преломления сильно зависит от температуры и длины волны света, при которой измеряют коэффициент преломления, то рефрактометрические определения проводят обычно при длине волны 589,3 нм (дублет при 589,0 и 589,6 нм) линии D в спектре излучения натрия и при строго постоянной температуре – чаще всего при 20±0,3˚С (требуется термостатирование). Полученные таким путем значения показателя преломления nD20 используют для нахождения фактора F по формуле (8.16) и содержания X определяемого вещества в анализируемом растворе.
Величину X определяют либо методом градуировочного графика, либо расчетным путем по формуле (8.15), либо линейной интерполяцией с использованием специальных рефрактометрических таблиц, если таковые имеются.
При использовании метода градуировочного графика измеряют показатели преломления n нескольких эталонных растворов с точно известным содержанием X определяемого вещества и по полученным данным строят градуировочный график в координатах n-X. Затем строго в тех же условиях измеряют показатель преломления анализируемого раствора и по градуировочному графику находят содержание определяемого вещества в анализируемом растворе.
Как было указано выше, расчет содержания X определяемого вещества по формуле (8.15) с использованием фактора F возможен только для интервала изменения X, в котором наблюдается линейная зависимость между X и показателем преломления. Чем меньше интервал между значениями n1 и n2, в котором лежит величина показателя преломления n анализируемого раствора, тем точнее расчет X по формулам (8.15) и (8.16).
Оптимальный диапазон изменения значений nD20 , при которых проводят количественные определения, составляет 1,3-1,7. Абсолютная ошибка определения показателя преломления не должна превышать ±(0,0002-0,0005), а для лекарственных препаратов – не более ±0,0002.
Рефрактометрический метод применяют при содержании в растворе определяемых веществ X˃1%.
Метод прост по выполнению, однако обладает невысокой точностью и чувствительностью.
Рефрактометрия используется при определении содержания многих веществ в растворах, например, органических жидкостей в водных растворах и в смесях, солей в водных растворах – хлорида, бромида, иодида натрия, бромида и иодида калия, хлорида кальция, гидрокарбоната, тиосульфата, цитрата, салицилата натрия, сульфата магния, лекарственных препаратов – амидопирина, аскорбиновой кислоты, гексаметилентетрамина, глюкозы, диэтиламида никотиновой кислоты в кордиамине, кодеина фосфата, кофеинбензоата натрия, новокаина, сульфацила растворимого, эфедрина и др.
Поляриметрия. Сущность метода и его применение в качественном анализе описаны в книге 1 (гл. 20, раздел 20.2.5). Использование поляриметриив количественном анализе основано на существовании зависимости между углом α оптического вращения плоскости поляризации монохроматического света при его прохождении через раствор, содержащий оптически активное вещество, и концентрацией c этого вещества в растворе. В определенном интервале изменения концентрации раствора наблюдается прямая пропорциональная зависимость (8.17) между углом оптического вращения и концентрацией раствора:
C = 100 α / ([α]l), (8.17)
Где [α] – удельное вращение, l – толщина слоя оптически активной среды, через которую проходит световой луч.
Удельное вращение – константа при постоянных температуре и длине волны света, характеризующая данное оптически активное вещество в растворе. Она остается постоянной в некотором интервале изменения концентрации оптически активного вещества в растворе данного растворителя. Для этого интервала и можно использовать формулу (8.17).
Обычно измерения проводят при температуре 20˚С и длине волны линии D в спектре излучения натрия (хотя это и не обязательно).
Поляриметрия применяется преимущественно в качественном анализе, например, при идентификации веществ – для доказательства их подлинности по величине удельного вращения. В количественном анализе метод используется, в частности, для определения содержания сахаров в растворе.
Эмиссионный спектральный анализ. Этот метод – один из старейших и детально разработанных оптических методов исследования и анализа. Сущность его изложена в книге 1 (гл. 20, раздел 20.2.1). Применение метода в количественном анализе основано на существовании симбатности между интенсивностью линий в спектрах излучения атомов, входящих в состав определяемого вещества, после его атомизации в источнике возбуждения ( пламя, электрическая дуга, электрический искровой разряд и др.) и содержанием определяемого вещества в анализируемом объекте: чем выше это содержание, тем больше излучающих атомов в плазме и тем интенсивнее излучение.
В узком интервале изменения содержания с определяемого вещества в анализируемом объекте (твердые фазы, растворы) интенсивность излучения I после атомизации пробы прямо пропорциональна концентрации:
I=kc, (8. 18)
Где коэффициент пропорциональности k, зависящий от природы анализируемого объекта и от условий проведения эксперимента, определяется эмпирически на основании измерения интенсивности излучения (после атомизации) эталонных проб с точно известным содержанием определяемого компонента.
Однако в более или менее широком интервале изменения содержания определяемого компонента в анализируемом объекте прямая пропорциональная зависимость между интенсивностью излучения и концентрацией не соблюдается (см. рис. 8.24).
Интенсивность I регистрируют фотометрически. Излучение от источника возбуждения после прохождения через конденсоры попадает в приемник излучения спектрального прибора – на фотоэлементы, фотоумножители (фотометрический способ регистрации) – или на фотопластинку (фотографический способ регистрации).
При фотометрическом способе регистрации энергия светового потока преобразуется в фототок, который затем усиливается и регистрируется в виде показаний на шкале прибора.
При фотографическом способе регистрации после проявления фотопластинки получают изображение в виде линий эмиссионного атомного спектра. Чем выше интенсивность излучения, тем интенсивнее и изображения линий на фотопластинке. Интенсивность линий на фотопластинке либо оценивают визуально (с невысокой точностью), либо измеряют на микрофотометре.
Обычно концентрацию определяемого вещества в анализируемом объекте находят с использованием способа градуировочных графиков. Для этого готовят несколько эталонных образцов, содержащих точно известное количество определяемого вещества, и измеряют интенсивность излучения после их атомизации. По полученным данным строят градуировочный графи в координатах показания прибора (пропорциональные интенсивности излучения) – содержание определяемого вещества в эталонном образце. Затем строго в таких же условиях проводят атомизацию с возбуждением спектра излучения анализируемого образца и измеряют показания прибора для этого образца, после чего по градуировочному графику находят содержание определяемого вещества в анализируемом объекте.
Абсолютная интенсивность линий в спектрах излучения атомов зависит от состава анализируемого объекта и от условий проведения анализа, поэтому все измерения для эталонных образцов и для анализируемой пробы должны проводиться в идентичных условиях. Содержание всех компонентов в эталонных образцах должно приближаться к содержанию тех же компонентов в анализируемой пробе.
Так как абсолютная интенсивность излучения зависит от целого ряда факторов, то был разработан метод определения относительной интенсивности (т. е. отношения интенсивностей) линии в спектре определяемого вещества в сравнении с интенсивность линии в спектре излучения эталона с точно известным содержанием эталонного вещества. Такой метод называют методом внутренних эталонов (внутренних стандартов), или методом гомологической (аналитической ) пары линий. В качестве эталона может служить либо элемент, содержащийся в самом анализируемом объекте, либо прибавляемый в точно известном количестве к эталонным образцам и к анализируемой пробе.
Так определяют, например, следовые количества магния в растворе с использованием молибдена (вводимого в виде молибдата аммония) в качестве эталона.
Для определения содержания следов магния в анализируемом растворе готовят серию эталонных растворов хлорида магния с содержанием магния в интервале 0,0001-10,0 мг/л, в каждой из которых прибавляют молибдат аммония до одинакового содержания молибдена 20,0 мг/л. В анализируемый раствор, содержащий магний в кол-ве от 0,0001 до 10,0 мг/л, так же прибавляют молибдат аммония до точно такого же содержания молибдена – 20,0 мг/л. Затем по специально разработанной методике атомизируют в искровом разряде между медными электродами эталонные растворы и анализируемый раствор и измеряют для всех проб интенсивность гомологической пары линий – магния при 2798,1 А и молибдена при 2816,2 А.
Строят градуировочный график, откладывая по оси ординат отношения интесивностей линийIMgIMo,а по оси абсцисс – содержание магния.
По полученному графику находят содержание магния в анализируемом растворе.
Методика позволяет определять до 0,01 мкг/мл магния в растворе. Ошибка определения составляет 5-10%.
Метод эмиссионного анализа высоко чувствителен – позволяет определять до -0,001 мкг/мл вещества в растворе. Ошибка метода достигает нескольких процентов: чаще всего -1-4% но может быть и выше.
Эмиссионная спектроскопия применяется в количественном анализе металлов, сплавов, руд и различных материалов, растворов неорганических, комплексных, металлоорганических соединений, сточных вод, для определения натрия, калия, магния, кальция в клинических пробах, в биологических жидкостях.
Атомно-абсорбционная пламенная фотометрия (спектрометрия). Принцип метода кратко описан в книге 1 (гл.20, раздел 20.2.2). Его применение в количественном анализе основано на существования зависимости между ослаблением интенсивности резонансного излучения (определяемого элемента), проходящего через пламя, в которое вводится определяемый элемент в виде его соединения и атомизируется, и концентрацией данного элемента в атомизируемой пробе.
Для поглощения плазмы в пламени справедлив основной закон светопоглощения
I = I0e-abl,
Где I0- интенсивность резонансного излучения (определяемого элемента), попадающего в плазму пламени; I –интенсивность ослабленного резонансного излучения (определяемого элемента), прошедшего через плазму пламени; а – коэффициент; b – концентрация в плазме пламени частиц, поглощающих резонансное излучение; l - толщина зоны плазмы пламени, в которой находятся поглощающие частицы.
Оптическая плотность
А = lg (I / I0) = abl
пропорциональна величине b. Можно полагать что, в свою очередь, концентрация bв пламени частиц, поглощающих резонансное излучение, прямопропорциональна концентрации с определяемого вещества в анализируемом растворе, вводимым в пламя:
b=k1c
Где k1 – коэффициент пропорциональности. Тогда:
A=ak1lc.
Обозначив k=ak1l и полагая l =const, можем представить формулу для оптической плотности в виде (8.19) :
A=kc. (8.19)
Прямая пропорциональная зависимость обычно выполняется лишь в определенном интервале изменения концентрации с.
Определение значения оптической плотности проводят измеряя интенсивность I0 излучения, прошедшего через пламя, не содержащее атомизируемой пробы (контрольное измерение), и интенсивность I излучения, прошедшего через плазму пламени с атомизируемой пробой. Используют как однолучевые так и двухлучевые атомно - абсорбционные спектрофотометры.
В качестве источника возбуждения резонансного излучения применяют набор ламп с полным катодом, содержащих определяемые элементы, излучение которых (свечение) инициируется в этих лампах.
Определение концентрации проводят с использованием методов градуировочных графиков, стандартных добавок, ограничивающих растворов.
В методе градуировочных графиков, как обычно, готовят серию эталонных растворов с точно известной концентрацией определяемого элемента, атомизируют в пламени пробу каждого эталонного раствора, измеряют оптическую плотность плазмы каждой пробы и по полученным данным строят градуировочный график в координатах оптическая плотность – концентрация определяемого элемента в эталонном растворе. После этого измеряют оптическую плотность плазмы, полученной при атомизации пробы анализируемого раствора, и по градуировочному графику находят концентрацию определяемого вещества в анализируемом растворе.
В методе ограничивающих растворов готовят два эталонных раствора с известными концентрациями с1 и с2 (с1 < с2) определяемого вещества, между которыми лежит концентрация с определяемого вещества в анализируемом растворе. После этого измеряют оптическую плотность А1, А2и Ах для всех трех проб и рассчитывают концентрацию сх способом линейной интерполяции:
cx = c1 + ( c2 – c1 ) (Ax – A1) / (A2 – A1 ).
Чем меньше интервал с2-с1тем точнее определение концентрации сx.
Чувствительность и погрешность метода атомно-абсорбционной пламенной фотометрии примерно такие же как и в методе эмиссионной пламенной фотометрии. Предел обнаружения составляет ≈ 10-12 -10-14 г, воспроизводимость анализа - около (1-4)%.
Метод применяется для определения более 70 элементов – больше, чем в случае эмиссионной пламенной фотометрии – в анализируемых объектах самой различной природы: в металлах и сплавах, в рудах, в минералах, в различных материалах, в лекарственных препаратах и т. д.
Инфракрасная спектроскопия. Основы метода изложены в книге 1 (гл.20, раздел 20.2.4). Количественный анализ в ИК в области спектра проводят так же как и при спектрофотометрии в УФ и в видимой области спектра, с использованием кювет с окошками из КВr, NaCl, LiF, CaF2 и других подходящих материалов. Водные растворы, как правило, не анализируют, так как, во-первых, сама жидкая вода поглощает ИК излучение и, во-вторых, пластинки из КВr и NaCl, чаще всего используемые в качестве материалов для окошек кювет, растворяются в воде. Поэтому обычно анализируют растворы определяемых веществ в неводных органических растворителях, часто - в тетрахлориде углерода, поскольку он в силу высокой симметрии молекулы имеет небольшое число полос и ИК спектре поглощения.
Разработаны методики количественного анализа различных веществ методами ИК спектрофотометрии.
Методы нарушенного полного отражения (НПВО) и многократно нарушенного полного внутреннего отражения (МНПВО). Эти методы применяются преимущественно (но не обязательно) в ИК спектроскопи и при исследовании и анализе тонких пленок, поверхностных слоев, сильно поглощающих и сильно рассеивающих сред, т. е. тогда, когда получение ИК спектров поглощения традиционными способами невозможно или проблематично. Однако, их можно использовать и при работе в УВИ области спектра.
Метод НПВО был разработан Дж. Фаренфортом в 1960г., а метод МНПВО – Н. Харриком в том же 1960г., хотя физические основы явления НПВО БЫЛИ ИЗВЕСТНЫ ДАВНО.
Принцип методов состоит в следующем. Пусть на границу раздела двух оптических сред с показателями светопреломления n1 и n2 падает луч света с интенсивностью I0 причем n1 > n2, т. е. луч света сначала проходит через прозрачную, оптически более плотную среду с показателем преломления n1 и затем отражается от поверхности прозрачной, оптически менее плотной среды с показателем преломления n2 . Если при отражении светового луча его спектральный состав и интенсивность не изменяются (I0=I), то такое явление называют полным внутренним отражением – граница раздела двух оптических сред ведет себя как идеальное зеркало.
Однако, падающий световой луч способен проникать на некоторое расстояние dв глубь оптически менее плотной среды и только после этого отражаться, повторно проходя расстояние dв оптически менее плотной среде. В целом, получается, что луч света проходит через оптически менее плотную среду с толщиной поглощающего слоя 2d.Величина dназывается глубина проникновения, или эффективная толщина поглощающей среды. Обычно она очень мала – соизмерима с длиной волны отражаемого света.
Если при прохождении светового луча через оптически менее плотную среду световая энергия частично избирательно поглощается, то спектральный состав отраженного света будет отличаться от спектрального состава падающего светового луча, а его интенсивность уменьшится: l=l0. Такое явление называется нарушенным полным внутренним отражением (НПВО). Регистрируя на спектрофотометре интенсивность отраженного излучения в определенном спектральном диапазоне (в ИК или УВИ области спектра), можно получить спектр поглощения тонкого приповерхностного слоя оптически менее плотной среды.
Если нарушенное полное внутренне отражение осуществляется многократно, то такое явление называют многократно нарушенным полным внутренним отражением(МНПВО). При МНПВО интенсивность полос поглощения в полученном спектре многократно увеличивается по сравнению с НПВО; сами спектры получаются более четкие.
Для НПВО и МНПВО справедлив основной закон светопоглощения, который можно представить в форме
R=I/I0=e-acNd, (8.20)
Где R – коэффициент отражения, α – показатель поглощения, с – концентрациясветопоглощающих частиц, N - число отражений, d –глубина проникновения (эффективная толщина светопоглощающей среды).
Глубина проникновения зависит от отношений показателей преломления n1/n2 , угла падения θ, длины волны падения падающего (отражаемого) света.
В элементах НПВО и МНПВО в качестве оптически более плотных сред применяют различные прозрачные материалы с высокими показателями преломления n1 –плавленый кварц (SiO2), сапфир (Al2O3), хлорид серебра, германий, кремний, смешанные кристаллы КРС-5 (TiBr+ТП), КРС-6 (TlCl+TiBr), жидкостные оптические элементы (например йодистый метилен CH2I2) и др. Разработаны и широко используются различные приставки НПВО и МНПВО к серийным промышленным спектрофотометрам.
Методы НПВО и МНПВО применяются для исследования и анализа тонких, в том числе мономолекулярных (при числе отражений N>500), слоев, водных растворов, ИК спектры поглощения которых труднодоступны при традиционных способах их получения, пленок, сильно поглощающих объектов, фармацевтических препаратов, при изучении состояния веществ вблизи границы раздела фаз электрод-раствор и т. д.
Нефелометрический анализ (нефелометрия). Метод основан на использовании зависимости между интенсивностью света, рассеиваемого частицами дисперсной системы, и числом этих частиц. При прохождении светового потока через светорассеивающую среду частицы этой среды рассеивают свет в различных направлениях с той же длиной волны, что и длина волны падающего светового потока. Если размеры Rсветорассеивающих частиц меньше длины волны λ рассеваемого света ( R < 0,1 λ ), то такое светорассеяние называют рэлеевским рассеянием (в отличиеот комбинационного рассеяния света, когда длины волн падающего и рассеянного излучения неодинаковы, или от эффекта Тиндаля, когда свет той же длины волны рассеивается более крупными частицами, чем при рэлеевском рассеянии света).
В нефелометрии интенсивность рассеянного света наблюдают (измеряют) в направлении, либо перпендикулярном, либо под каким-то углом по отношению к направлению падающего светового потока. Обычно наблюдения и измерения ведут в направлении перпендикулярном к направлению распространения падающего света.
Интенсивность рэлеевского рассеяния света зависит от природы рассеивающей среды, размеров частиц и их числа, показателей светопреломления частиц и среды, длины волн и интенсивности падающего света, угла рассеивания. При размерах частиц, существенно меньших длины волны λ падающего света (меньше 0,1 λ), интенсивность рассеянного света обратно пропорциональна четвертой степени длины волны и описывается уравнением Рэлея:
I = I0FV2 (1 + cos2θ)N / λ 4R2 (8.21)
Где I и I0- соответственно, интенсивность рассеянного и падающего света с длиной волны λ; F – функция, зависящая от показателей преломления частиц дисперсной фазы и дисперсионной среды; V – объем частицы, принимаемой за сферическую; θ – угол между направлениями падающего и рассеянного света; N – общее число частиц в рассеивающей среде; R – расстояние от рассеивающей частицы до приемника рассеянного излучения (до наблюдателя).
Если нефелометрические измерения проводить в условиях, когда величины F, V, λ, ϴ и R остаются постоянными, то, тогда уравнение можно представить в виде:
I=I0kN, (8.22)
Где коэффициент пропорциональности k определяется эмпирически. В таком случае в соответствии с формулой интенсивность рассеянного света пропорциональна числу рассеивающих частиц N, а при постоянном объеме рассеивающей среды – их концентрации. Соотношение и лежит в основе нефелометрических определений.
Измерения проводят с использованием специальных приборов – нефелометров или флуориметров.
Концентрацию определяемого вещества находят либо методом градуировочного графика, построенного на основании измерения интенсивности рассеяния эталонных проб с точно известной концентрациейопределяемого вещества, либо с использованием стандарта. В последнем случае измеряют отношение интенсивностей светорассеяния стандартного образца с точно известной концентрацией определяемого вещества и анализируемой пробы:
Is = I0kNs, Ix=I0kNx, Ix /Is=Nx/Ns,
Где символы «s» и «х» относятся к стандартному и анализируемому образцам соответственно.
При одинаковом объеме проб стандартного и анализируемого образцов отношение чисел рассеивающих частиц Nx/ Ns равно отношению их концентраций сх и сs:
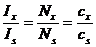
Отсюда:
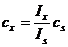 .
.
Найдя cx и измерив отношение интенсивностей Ix/Is можно рассчитать концентрацию cx определяемого вещества в анализируемом образце.
Нефелометрию применяют при анализе тонких суспензий, эмульсий, коллоидных растворов и т. д. Например, нефелометрически можно определить сульфаты в водных суспензиях сульфата бария, хлориды в водных суспензиях хлорида серебра. Для стабилизации суспензий в них вводят добавки желатина.
Ошибки определения концентраций нефелометрическим методом составляют 2-5%.
Турбидимитреческий (фототурбидиметрический) анализ (турбидиметрия, фототурбидиметрия). Метод основан на использовании зависимости между ослаблением интенсивности светового потока, проходящего через светорассеивающую среду, за счет рассеивания света частицами этой среды, и их концентрацией. При турбидиметрических измерениях через светорассеивающую среду пропускают световой поток с интенсивностью I0 , измеряют его интенсивность I после прохождения им светорассеивающей среды. При наличии частиц, рассеивающих свет (рэлеевское рассеяние), очевидно, что I<I0. В таком случае справедливо соотношение
Аналогичное соотношение для основного закона светопоглощения. В выражении величину S, играющую роль оптической плотности, иногда называют мутностью; k – коэффициент пропорциональности, зависящий от размера рассеивающих частиц, длины волны падающего света, коэффициентов светопреломления частиц и среды; c – концентрация светорассеивающих частиц; l – толщина рассеивающего слоя; r -= kc – коэффициент мутности, иногда называемый также мутностью.
Уравнение предполагает, что соблюдается формула Рэлея и размер частиц Rменьше длины волны λ падающего света (R≤ 0.1λ).
Для измерения мутности S используют обычные фотоэлектроколориметры, а также специальные приборы – турбидиметры.
Метод обладает меньшими чувствительностью точностью, чем нефелометрия. Ошибка определения концентрации турбидиметрическим методом составляет около 5%.
Как и нефелометрию, турбидиметрию можно использовать для определения сульфатов (по суспензии сульфата бария), хлоридов ( по суспензии хлорида серебра) и др. веществ, используя для нахождения содержания определяемого вещества метод градуировочного графика.
