
Партнерка на США и Канаду по недвижимости, выплаты в крипто
- 30% recurring commission
- Выплаты в USDT
- Вывод каждую неделю
- Комиссия до 5 лет за каждого referral
Хроматографическое разделение a, b-аномеров ацилированного dA-8-13С
,
РГАУ - МСХА им. , кафедра неорганической и аналитической химии
В современных физико-химических исследованиях ЯМР-спектроскопия является одним из основных методов определения детальной структуры РНК и ДНК в растворе. Эти исследования требуют компонентов нуклеиновых кислот (КНК), обогащенных изотопами 13С и 15 N.
Одним из востребованных соединений является 2¢-дезоксиаденозин-8-13С (dA-8-13С) и его a-D-аномер. Для получения этих соединений нами разработан метод прямой конденсации пуриновых производных с галогенацилпентозами в присутствии молекулярных сит. Идея метода заимствована из работы [1]. Реакцию конденсации проводили в безводном дихлорметане в присутствии молекулярных сит 5 Å 72 часа при комнатной температуре по схеме:
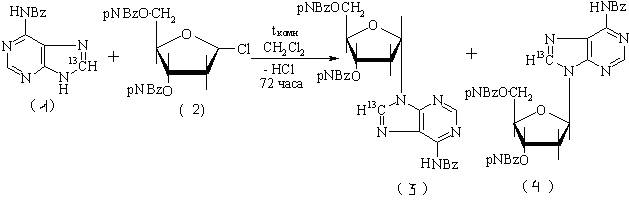
где pNBz – п-нитробензоил; (1)- 6-бензоиладенин-8-13С;(2)- 1-хлор-2-дезокси-3,5-ди-О-п-нитробензоил-D-рибоза; (3) - a-защищенный dA- 8-13С (4) - b-защищенный dA-8-13С.
Найдены условия выделения продукта из реакционной смеси, его дальнейшей очистки и снятия защиты п-нитробензоильных групп, утилизации меченого сырья, повышающие выход меченого продукта.
В работе ставилась задача разделения a, b-аномеров ацилированного dA и был применен и отработан более эффективный вариант разделения методом колоночной хроматографии по сравнению с фракционной кристаллизацией предложенной в работе [1].При хроматографическом способе выделения общий выход ацилированного меченного dA (a, b-аномеров) составлял 41 %, считая на ацилдезокси-рибозу (2) против 26%, полученного по методике [1].
Хроматографическое разделение проведено на колонке с силикагелем L 100/400 (²Хемапол²) (70х2,3) в два этапа. Для каждого этапа подобраны оптимальные условия элюирования. Сначала проводили разделение ацилированного dA (3 и 4) от исходной непрореагировавшей ацилдезокси-рибозы (2). Элюентом были 3 последовательных раствора: толуол; этилацетат-толуол (10:1); этилацетат (продвижение зоны контролировалось визуально). Фракции анализировались методом ТСХ (этилацетат-толуол 3:2), проявление солянокислым цистеином при 80 °С (Rf защ. dA 0,216; Rf 1 защ. дез.-риб. 0,93; Rf2 0,705). При элюировании толуол пропускался до полного вымывания ацилдезокси-рибозы (продвижение желтой зоны), далее начиналось элюирование продукта смесью этилацетат-толуол (10:1) (продвижение бесцветной зоны), для окончательного элюирования продукта пропускали чистый этилацетат.
На втором этапе проводилось разделение ацилированного dA на a, b-аномеры. Синтезированный дезоксиаденозин содержал 2 аномера (a, b), а природные рибонуклеозиды и дезоксирибонуклеозиды имеют b-конфигурацию гликозидного центра. В связи с этим, конечная стадия синтеза ацилированного dA-8-13С должна предполагать разделение a, b-аномеров и получение природного аналога. Для выбора элюирующей системы проводились исследования методом ТСХ. Для повышения эффективности разделения проб на пластинах применен способ многократного хроматографирования на одной силуфольной пластине.
Исследовались смеси этилацетат-толуол в различных соотношениях, оптимальной оказалась система этилацетат-толуол (3:2), после 3-х-кратного хроматографирования в которой наблюдалось деление ацил-dA на a, b-аномеры. Препаративное разделение ацил-dA проводилось на колонке с силикагелем L100/400 (70х2,3) смесью этилацетат-толуол (3:2), фракции анализировались ТСХ (этилацетат-толуол 3:2, Rf b-защ. dA 0,115; Rf a-защ. dA 0,098).
При данных условиях хроматографического разделения из смеси аномеров ацил-dA получали 33 % ацил-b-dA, 15 % a-аномера и 27 % исходной смеси a, b-dA, соотношение b/a превышало 2, что благоприятно для получения природного аналога b-dA.
Проведен ПМР анализ a - и b-2'-дезоксиаденозина-8-13С (3 и 4), защищенного в положении 3 и 5 2-дезокси-рибозы п-нитро-бензоильной группой и в положении 6 аденина бензоильной группой. На рис.1 приведен фрагмент ПМР спектра, по которому возможно судить о положении и концентрации изотопа 13С.
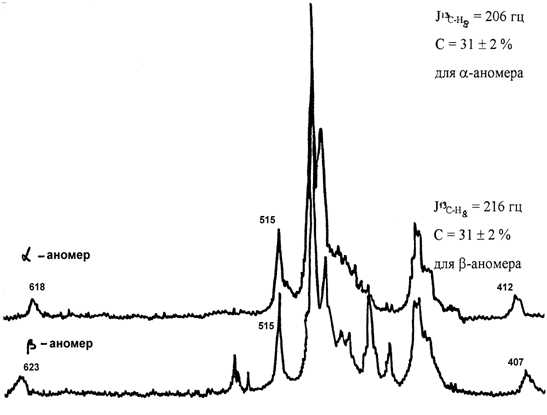
Рис.1.Спектр ПМР a - и b-ацилированного 2¢-дезоксиаденозина-8-13С, снятый на
ЯМР-спектрометре “Тесла” (60 МГц), пробы растворялись в (CD3)2CO.
Завершающая стадия состояла в снятии защиты п-нитробензоильных и бензоильной групп в ацил- dA. Были исследованы способы снятия защиты у ацилированного dA обработкой его метанольного раствора 0,1 М раствором метилата бария по методике [1], раствором этилата бария и обработкой газообразным аммиаком при 50°С в течение 24 часов. Последний способ был предпочтительнее. После снятия защиты выход 2'-дезоксиаденозина-8-13С составлял 28 %, его a-аномера - 12 %. Анализ ТСХ (н-бутанол-насыщенный NH3), проявление солянокислым цистеином при (80-85 °C) дал Rf dA 0,535 (что совпадает с эталоном dA). УФ-спектр: lmax =258 нм. На рис. 2 приведен обзорный спектр ЯМР-13С dA-8-13C.
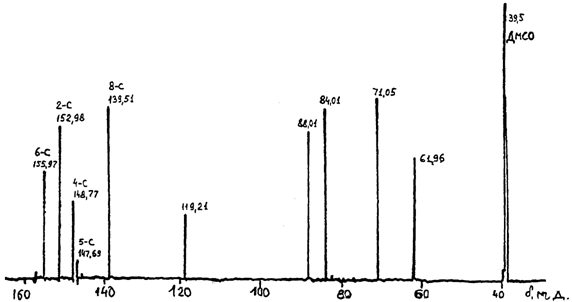
Рис. 2 Обзорный спектр ЯМР-13С dA-8-13C.
Сигналы атомов углерода ароматической части лежат в области 160-139 м. д. (d6-С = 155,97; d2-С = 152,29; d4-С = 148,77; d5-С = 147,69; d8-С = 139,51). Сигналы атомов углерода рибозной части лежат в области 120-60 м. д. (d1 = 119,21; d2 = 88,01; d3= 84,01; d4 = 71,05; d5 = 61,96 м. д.). Из соотношения интенсивностей сигналов изотопов 8-12С и 8-13С (![]() n = 15,8 Гц) определена концентрация изотопа 8-13С [% 13С = 307,74/(178,22 + 307,74) = 63 %].
n = 15,8 Гц) определена концентрация изотопа 8-13С [% 13С = 307,74/(178,22 + 307,74) = 63 %].
Проведен также ПМР анализ dA-8-13C (Varian XL-400, DMSO) (рис.3). Так как сигнал 8-протона в дезоксинуклеозиде расположен в слабопольной области (d8-Н = 8,35 м. д.) и не перекрывается с сигналами других протонов (рядом находится сигнал 2-Н с d2-Н = 8,15 м. д.), сателлиты от взаимодействия с ядром 13С легко определяются (d1 = 8,6 и d2 = 8,1 м. д., J C - 8-H = 212 Гц) и концентрация 13С рассчитывается по отношению суммы интенсивностей сигналов сателлитов к общей интенсивности сигналов 8-Н и сателлитов (62 % 13С).
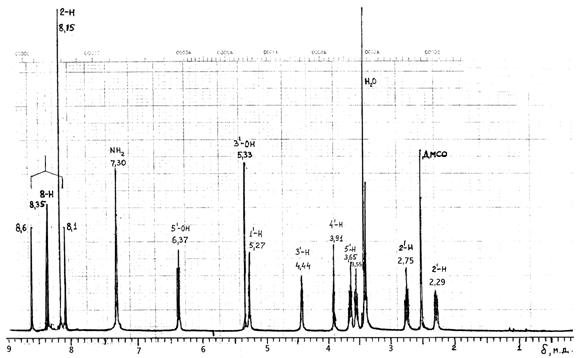
Рис. 3 Обзорный спектр ПМР dA-8-13C, снятый на спектрометре Varian-XL-400, DMSO.
Сигнал 6-NH2-группы в виде широкого синглета имеет химический сдвиг 7,30 м. д. Сигналы протонов рибозной части прописаны с высоким разрешением каждый в отдельности. Сигнал ОН при 5¢-С с химическим сдвигом 6,37 м. д. расщепляется на 2 дублета с J1 = 7,88 Гц и J2 = 6,06 Гц. Сигнал ОН при 3¢-С с d = 5,33 м. д. расщепляется на дублет с J = 3,99 Гц, сигнал Н при 1¢-С с d =5,27 м. д. расщепляется на J1 = 6,74 Гц и J2 = 4,92 Гц (рис.4). Сигнал Н при 3¢-С с d = 4,44 м. д. расщепляется на дублет дублетов дублетов с J1 = 2,91 Гц; J2 = 6,72 Гц и J3 = 4,31 Гц. Сигнал Н при 4¢-С с d = 3,91 м. д. расщепляется на дублет триплетов дублетов с J1 = 2,52 Гц; J2 = 4,24 Гц и J3 = 6,76 Гц. Сигнал двух протонов при 5¢-С представлен в виде дублета дублетов с J1 = 11,87 Гц и J2 = 9,08 Гц при d1 = 3,65 м. д. дублета дублетов дублетов с J1 = 6,64 Гц; J2 = 6,79 Гц и J3 = 9,48 Гц при d2 = 3,55 м. д. Сигнал двух протонов при 2¢-С расщепляется на дублет дублетов и дублетов с J1 = 5,63 Гц; J2 = 2,83 Гц и J3 = 13,46 Гц при 2,75 м. д. и на дублет дублетов дублетов с J1 = 2,91 Гц; J2 = 6,06 Гц и
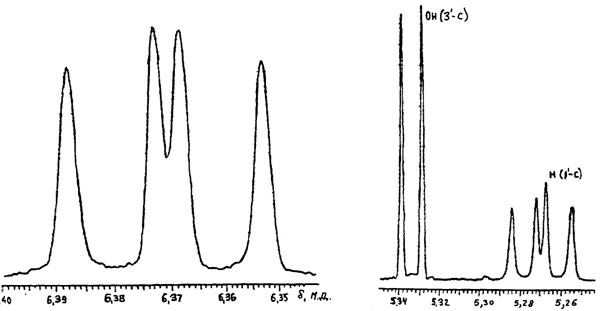
ОН при 5'-С ОН (3'-С) Н (1'-С)
Рис.4. Фрагменты спектра ПМР dA-8-13C.
J3 = 13,06 Гц при 2,29 м. д.
Отметим достоинства метода конденсации 6-бензоиладенина с 1-хлор-2-дезокси-3,5-ди-О-п-нитробензоил-D-рибозой в присутствии молекулярных сит (5Å): а) синтез осуществляется в мягких условиях, дает наиболее чистые продукты с высокими выходами и соотношением b/a аномеров – 2, что благоприятно для получения 2¢-dA-8-13C (b-аномера);
б) возможна наиболее полная утилизация меченого исходного сырья: бензоиладенина-8-13С и немеченой ацилированной 2-дезокси-D-рибозы. При этом выход ацил-2¢-dA-8-13C (3 и 4) повышался до 60-75 %, а после снятия защиты и перекристаллизации выход 2¢-dA-8-13C (b-аномера) достигал 30 % и 15 % для a-аномера.
Выход продукта можно повысить проведя повторное хроматографирование неразделенных фракций a, b-аномеров.2'-dA-8-13C. Неполное разделение a, b-аномеров ацил-dA было устранено повторным разделением смеси аномеров, которую присоединяли к последующим операциям разделения.
Проводился поиск хроматографических систем, улучшающих качество разделения a, b-аномеров (3 и 4). Целью было улучшение разделительной способности системы этилацетат-толуол введением полярных растворителей.
В чистом этилацетате происходило частичное разделение a, b-аномеров ацил-dA, но пятна были сильно размытыми. В толуоле пятна практически не двигались со старта. Оптимальное соотношение этилацетат-толуол было 3:2. Для улучшения разделения применяли добавки, не ускоряющие движение пятен, но улучшающие динамические характеристики системы: уменьшение продольной диффузии и обеспечение равновесности процесса между двумя фазами. Такой добавкой могли быть типичные полярные вещества: ацетонитрил, спирты, кислоты, которые были проверены для улучшения разделения a, b-аномеров.
Наилучшими оказались метиловый спирт и муравьиная кислота при соотношении компонентов 20:20:1. В этой системе разделение осуществлялось за одно хроматографирование по сравнению с исходной системой, в которой разделение достигалось только при трехкратном хроматографировании. Разделительная способность этой системы характеризуется максимумом (рис.5). Аналогичные результаты по разделению аномеров получены в системе этилацетат-циклогексан с добавкой муравьиной кислоты (20:20:1), (рис.6).
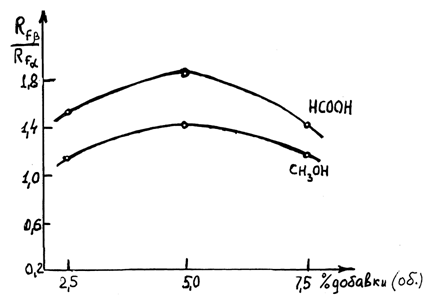
Рис. 5. Зависимость эффективности разделения a, b-аномеров ацилированного dA (3 и 4) от % добавки муравьиной кислоты и метилового спирта в системе этилацетат-толуол 1:1.
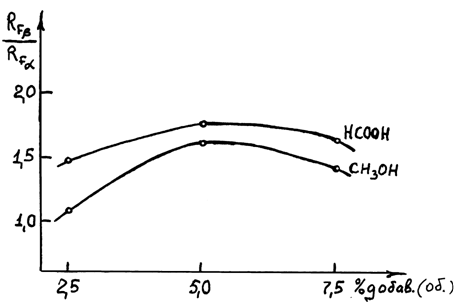
Рис.6. Зависимость эффективности разделения a, b-аномеров ацилированного dA (3 и 4) от % добавки муравьиной кислоты и метилового спирта в системе этилацетат-циклогексан 1:1.
Результаты разработки хроматографического разделения a, b-аномеров (3 и 4) представлены в табл. 1. Эффективность разделения на колонке достигала 62-65 % (в случаях 1, 3 ,4,5) и в менее удачном эксперименте (2) была 47-50 %.
Таблица 1.
Хроматографическое разделение ацилированного dA* (3 и 4) на a, b-аномеры на силикагеле типа L 100/400 (²Хемапол²).
Элюент этилацетат-толуол (3:2).
п/п | Высота слоя и диаметр, мм | Количество наносимой пробы ацил - dA*, г | Количество выделенного b-аномера ацил-dA* , г (%) | Количество выделенного a-аномера ацил-dA*, г (%) | Количество неразделенных a, b-аномеров ацил-dA*, (%) | Соотношение b/a |
1 | 700х23 | 0,82 | 0,27 (33 %) | 0,12 (15 %) | 0,22 (27 %) | 2,2 |
2 | 0,50 | 0,12 (24 %) | 0,05 (10 %) | 0,19 (38 %) | 2,4 | |
3 | 700х23 | 0,64 | 0,235 (37 %) | 0,147 (23 %) | 0,23 (36 %) | 1,61 |
4 | 750х23 | 0,423 | 0,140 (33 %) | 0,063 (15 %) | 0,20 (47 %) | 2,2 |
5 | 750х23 | 0,621 | 0,205 (33 %) | 0,099 (16 %) | 0,164 (26 %) | 2,1 |
Обозначение: * - -8-13C.
Литература
1. Ness R. K. 2'-Deoxyadenosine and its a-D-anomer. A direct synthesis of anomeric purine 2'-deoxyribonucleosides // Synthetic Procedures in Nucleic Acid Chemistry, ed. Zorbach W. W., Tipson R. S., Interscience series. – 1968. – V.1. – P.183-187.
