

Партнерка на США и Канаду по недвижимости, выплаты в крипто
- 30% recurring commission
- Выплаты в USDT
- Вывод каждую неделю
- Комиссия до 5 лет за каждого referral
Примечание: Биографические сведения авторов смотри в Бутлеровских сообщениях. 2002. Т.2. №6. 31.
Публикация доступна для обсуждения в рамках функционирования постоянно действующей интернет-конференции “Бутлеровские чтения”.
Поступила в редакцию 20 июля 2005 г. УДК 544.47+544.18.
Теоретический анализ адсорбции молекулярного кислорода на кластере серебра Ag4
1*+, 1,2, 1
1 Кафедра физической и коллоидной химии. Томский государственный университет,
пр. Ленина, 36 г. Томск 634050. Россия.
Тел. (3822) 42-07-80. E-mail: mihan555@yandex.ru
2 Кафедра органической химии. Томский государственный педагогический университет.
пр. Комсомольский, 75. г. Томск 634041. Россия.
Тел. (3822) 59-14-54. E-mail: *****@***edu. ru
_________________________________________________
* Ведущий направления; + Поддерживающий переписку
Ключевые слова: теория функционала плотности; B3LYP/DGDZVP; адсорбция; каталитические процессы.
Аннотация
В настоящей работе на уровнях теории HF/DGDZVP и B3LYP/DGDZVP выполнены квантово-химические расчеты энергий адсорбционного взаимодействия в системах, содержащих четырехатомный кластер серебра и молекулярный адсорбированный кислород. Показано, что уровень теории HF/DGDZVP позволяет получить качественные представления, хорошо согласующиеся с экспериментальными данными, однако не дает необходимой точности при описании адсорбционных взаимодействий, в то время как уровень теории B3LYP/DGDZVP позволяет адекватно оценивать геометрию адсорбированных форм и адсорбционные взаимодействия. Показано, что в окислительных каталитических процессах, протекающих на поверхности серебра, с большей вероятностью участвует молекулярный кислород в синглетном состоянии.
In the present work quantum-chemical calculations of adsorption interaction energies were carried out in the systems containing four-atom silver cluster as well as molecular adsorbed oxygen located at different adsorption sites with respect to the cluster surface. The calculations were performed at HF/DGDZVP and B3LYP/DGDZVP levels of theory. It was shown that the HF/DGDZVP level of theory allows obtaining a qualitative model correlating well with experimental data. However, this method does not provide an appropriate precision when describing adsorption interactions, while the B3LYP/DGDZVP provides an adequate accuracy. It was stated that molecular oxygen participates in oxidative catalytic processes, carried out over the silver-containing catalyst surface, in its singlet state.
1. Введение
Хорошо известно, что серебро является одним из наиболее активных катализаторов в процессах парциального окисления органических соединений. Среди подобных процессов можно отметить эпоксидирование этилена [1], окисление метанола в формальдегид [2], окисление этиленгликоля в глиоксаль [3], окислительная конверсия метана [4], эпоксидирование пропилена [5] и др. В ряде работ с использованием различных физико-химических [4, 6-17] и расчетных [16, 18-22] методов изучался механизм взаимодействия поверхности серебра с молекулярным и атомарным кислородом. Понимание сути происходящих при взаимодействии процессов важно, поскольку оно дает представления не только о пути образования на поверхности серебра адсорбированного атомарного кислорода, который является наиболее важным поверхностным интермедиатом в окислительных каталитических процессах, но и позволяет предсказывать поведение системы с адсорбированным атомарным кислородом в присутствии других компонентов процесса (газофазные молекулы, активные промежуточные частицы и др.).
С точки зрения детализации и интерпретации механизмов окислительных процессов с участием молекулярного кислорода важно иметь представление об энергиях адсорбционного взаимодействия участников процесса с активными центрами на поверхности серебросодержащего катализатора, а также о структуре и колебательных частотах промежуточных и переходных состояний, возникающих с их участием. Одним из способов исследования подобных взаимодействий являются теоретические квантово-химические расчеты с применением методов теории функционала плотности (DFT). Использование достаточно точных функционалов и базисных наборов позволяет получить адекватные значения необходимых физических параметров при относительно небольших затратах компьютерных и временных ресурсов. В мировой литературе представлено значительное количество работ, посвященных расчетам систем адсорбат/поверхность. В зависимости от целей расчетов применяются различные расчетные программы и коды (VASP [18], DACAPO [16], Gaussian [19], SIESTA [26], Turbomole [25], ADF [28], etc.).
Для исследования указанных выше особенностей окислительных процессов удобно использовать кластерные модели. Небольшие кластерные модели показали свою полезность при изучении адсорбции индивидуальных компонентов и обеспечивают достаточно точное описание адсорбированных структур, колебаний, а также энергий физической и химической адсорбции [23-26]. С использованием DFT методов показано, что оптимизированная структура четырехатомного кластера серебра, часто используемого для расчетов, является планарной и ромбической.
Рядом исследователей [27-29] показано, что одним из оптимальных с точки зрения затрат временных и компьютерных ресурсов методов для DFT расчетов систем, содержащих атомы элементов пятого периода (включая серебро), является сочетание гибридного трехпараметрического функционала B3LYP с полноэлектронным валентно-расщепленным базисом с добавлением поляризационных функций DGDZVP. Этот базисный набор специально оптимизирован для DFT расчетов соединений с тяжелыми атомами.
Несмотря на наличие большого количества экспериментальных и теоретических исследований взаимодействия серебра с кислородом до сих пор нет единой трактовки наиболее эффективных мест адсорбции молекулярного кислорода, а также структуры поверхностных форм. Кроме того, не во всех работах проводился учет спиновой природы молекулы кислорода. В связи с этим целью настоящей работы было исследование процессов адсорбции и диссоциации молекулы кислорода в синглетном и триплетном состояниях на небольшом четырехатомном кластере серебра на уровнях теории HF/DGDZVP и B3LYP/DGDZVP.
2. Метод расчета
В настоящей работе на уровнях теории HF/DGDZVP и B3LYP/DGDZVP выполнены расчеты систем, содержащих кластер серебра и газофазную молекулу кислорода в синглетном или триплетном состоянии. Расчеты выполнены в программном пакете Gaussian’03 [30, 31]. Исходное состояние серебра представляло собой четырехатомный кластер, имеющий плоскую геометрию. Расстояние между соседними атомами серебра в исходной структуре составляло 4,086 Å [32]. Для сравнения выполнен расчет исходного состояния кластера серебра, в котором атомы размещены в вершинах тетраэдра с теми же геометрическими параметрами. Молекула кислорода помещалась над поверхностью кластера в четырех положениях – «atop» (A), «bridge» (B), «hollow, above» (C), «hollow, in-plane» (D) (Рисунок 1). В исходных структурах адсорбаты в положениях «atop», «bridge» и «hollow, in-plane» размещались на расстояниях не менее 1.4 Å.
Геометрия всех полученных структур была полностью оптимизирована. Во всех случаях была проверена природа стационарной точки посредством расчетов колебательных частот. Большая часть оптимизированных структур обладала глобальным энергетическим минимумом и имела только действительные частоты. Термодинамические параметры рассчитываемых систем были скорректированы с учетом нулевой колебательной энергии и приведены к нормальным условиям (298.15 K, 1 атм) с использованием термических поправок к энтальпии и свободной энергии. Выполнен также расчет термодинамических параметров для температуры 830 K и давления 1 атмосфера согласно [33]. Отсутствие мнимых колебательных частот подтверждало основное состояние системы. Для случаев, когда структура имела мнимые частоты, проводился IRC расчет для определения наличия переходного состояния. Энергия адсорбции молекул кислорода на поверхности кластера определялась как разность полной энергии системы адсорбат/поверхность и суммы энергий изолированного адсорбата и кластера серебра.
3. Результаты и обсуждение
3.1.1. Кластер серебра Ag4. В Таблице 1 представлены результаты расчетов четырехатомного кластера серебра на уровнях теории HF/DGDZVP и B3LYP/DGDZVP. По результатам оптимизации структуры установлено, что кластер серебра имеет планарную ромбическую структуру. Длина связи между соседними атомами Ag при расчетах на уровне теории HF/DGDZVP составила 3.10 Å при значениях углов ромба 124.5o и 54.5o. Соответствующие показатели для уровня теории B3LYP/DGDZVP составили 2.91 Å, 124.1o и 55.9o. Результаты расчетов на уровне теории B3LYP/DGDZVP находятся в лучшем согласии с экспериментальными и расчетными результатами (например, расстояние Ag-Ag составляет 2.889 Å в [34], 2.838 Å в [24], 2.66 Å в [25]). Разница в геометрических параметрах может быть объяснена нейтральностью использованного в настоящей работе кластера серебра.
Таким образом, можно сделать вывод, что метод B3LYP/DGDZVP может быть использован для расчета серебросодержащих систем. Необходимо отметить, что в случае, когда исходный кластер серебра был представлен в виде тетраэдра, оптимизированная структура переходила в планарную ромбическую форму с теми же геометрическими параметрами, что и для плоской исходной геометрии, для обоих использованных уровней теории.
3.1.2. Молекула кислорода. Результаты расчетов показали сокращение длины связи O-O при сравнении экспериментальных и расчетных данных на уровне теории HF/DGDZVP и хорошее соответствие эксперимента и расчетов на уровне теории B3LYP/DGDZVP. На уровне теории HF/DGDZVP длина связи O-O составила 1.167 и 1.172 Å для синглетного и триплетного состояний, соответственно, при экспериментальном значении параметра 1.21 Å [35]. Метод B3LYP/DGDZVP обеспечивает хорошее соответствие рассчитанных (1.222 Å для обеих форм кислорода) и экспериментальных значений. По-видимому, это связано с большей точностью метода B3LYP, обусловленной учетом электронной корреляции. С использованием указанных уровней теории проведены расчеты энергий основного состояния синглетного и триплетного кислорода (Таблица 1). Наблюдаемые различия в значениях энергий синглетного и триплетного состояний (0.0849 a. u. для HF/DGDZVP и 0.061459 a. u. для B3LYP/DGDZVP) показывают, что энергетически более выгодным является триплетное состояние кислорода, что хорошо согласуется с современными представлениями (например, [36]).
3.2. Физическая адсорбция газофазных молекул синглетного и триплетного кислорода. Для расчетов на уровне теории HF/DGDZVP в качестве адсорбата выбран синглетный кислород, который в исходной структуре помещался поочередно во всех положениях, указанных на Рисунке 1. В Таблице 2 представлены геометрические параметры структуры, полученной при оптимизации исходных систем, содержащих четырехатомный кластер серебра и молекулярный кислород в положениях «atop», «bridge» и «hollow, in-plane» (структура 2a). Независимо от начального расположения атомов исходные состояния переходили в оптимизированную структуру 2a, которая являлась планарной и ромбической. Из таблицы видно, что атомы кислорода встраиваются между двумя соседними атомами серебра. В рассчитанных ИК спектрах всех структур наиболее интенсивная полоса соответствует колебаниям атомов кислорода по направлению к центру кластера. Значение длины связи Ag-O в оптимизированной структуре достаточно близко к экспериментальным значениям (согласно [37] этот параметр находится в интервале 2.17-2.42 Å).
Полученная структура согласуется с данными работы [4], в которой при изучении механизма диффузии кислорода на одиночном кристалле серебра и поликристаллическом образце показано, что объемный ![]() кислород в условиях реального каталитического процесса и температурах ниже 973 K может выходить к поверхности серебра и встраиваться между двумя соседними атомами. Это приводит к наведенной кислородом реструктуризации поверхности серебра. Кроме того, небольшое расширение решетки серебра при включении атомов кислорода, полученное нами (Таблица 2), подтверждает экспериментальные результаты [работа 7 и ссылки в ней]. Необходимо отметить, что в оптимизированной структуре расстояние между атомами кислорода составляет 1.484 Å, что свидетельствует о тенденции к диссоциации молекулы кислорода в указанных исходных структурах. Это подтверждено также результатами эксперимента [8-10].
кислород в условиях реального каталитического процесса и температурах ниже 973 K может выходить к поверхности серебра и встраиваться между двумя соседними атомами. Это приводит к наведенной кислородом реструктуризации поверхности серебра. Кроме того, небольшое расширение решетки серебра при включении атомов кислорода, полученное нами (Таблица 2), подтверждает экспериментальные результаты [работа 7 и ссылки в ней]. Необходимо отметить, что в оптимизированной структуре расстояние между атомами кислорода составляет 1.484 Å, что свидетельствует о тенденции к диссоциации молекулы кислорода в указанных исходных структурах. Это подтверждено также результатами эксперимента [8-10].
Оптимизированная структура исходного состояния, в котором молекулярный кислород находился в положении «hollow, above», представлена в Таблице 2 (структура 2b). Видно, что кластер серебра сохраняет планарную ромбическую структуру. При этом наблюдается изменение геометрических параметров кластера. Молекула кислорода ориентирована под углом к плоскости кластера, ее геометрические параметры соответствуют параметрам изолированной газофазной молекулы. Расстояние между ближайшими атомами серебра и кислорода превышает экспериментальные значения. Относительное сокращение длин связей r(Ag(3)-Ag(4)) и r(Ag(4)-Ag(1)) по сравнению с изолированным кластером серебра, по-видимому, связано с наведенным молекулой кислорода смещением плотности заряда от атома серебра (Ag(4)) по направлению к молекуле кислорода (малликеновские заряды на атомах серебра Ag(4) и Ag(2) составили 0.218 и 0.242 e, соответственно; заряды на атомах кислорода O(5) и O(6) составили -0.012 и 0.025 e, соответственно). Вектор заряда направлен от атома серебра Ag(4) к атому Ag(2). В рассчитанном ИК спектре наиболее интенсивная полоса соответствует диссоциативным колебаниям в молекуле кислорода.
Из Таблицы 1 видно, что полные энергии систем, переходящих в структуру 2a, практически одинаковы. Однако близость энергий состояний с различным начальным положением молекулы кислорода не позволяет сделать однозначный выбор наиболее благоприятного положения. В то же время, структура 2b обладает более высокой энергией, что свидетельствует о ее меньшей выгодности. При этом оцененные энергии взаимодействия позволяют говорить о слабом взаимодействии молекулы кислорода, расположенной в положении «hollow, above», с кластером серебра.
Таким образом, расчеты на уровне теории HF/DGDZVP качественно подтверждают экспериментальные данные о встраивании атомов кислорода в решетку серебра, однако не дают представлений о наиболее энергетически выгодном месте для адсорбции молекулы синглетного кислорода. Ввиду невысокой точности метода HF/DGDZVP при определении энергий взаимодействия, а также известных предположений о меньшей активности триплетного состояния молекулы кислорода, расчеты систем, содержащих триплетный молекулярный кислород, на этом уровне теории не проводились.
Более высокая точность уровня теории B3LYP/DGDZVP позволила использовать его для расчета исходных структур, в которых молекулярный кислород находился в триплетном состоянии. Результаты расчетов и оптимизации исходных структур, содержащих триплетный молекулярный кислород в положениях «bridge», «hollow, in-plane» и «hollow, above» представлены в Таблице 2 (структура 2c). Видно, что структура является планарной. Молекула кислорода находится в плоскости кластера, но на некотором расстоянии от него, и параллельна связи Ag(2)-Ag(3) для структуры «bridge», связи Ag(3)-Ag(4) для структуры «hollow, above» и Ag(1)-Ag(4) для структуры «hollow, in-plane». Оптимизированная структура исходной системы, содержавшей молекулу триплетного кислорода в положении «atop», имела строение, представленное в Таблице 2 (структура 2d). Значение длины связи r(O=O) в молекуле кислорода близко к параметрам газофазной молекулы.
Результаты расчетов показывают (Таблица 1), что системы, содержащие триплетный кислород, обладают более низкими значениями полных энергией по сравнению с системами, содержащими кислород в синглетном состоянии. Рассчитанные и оптимизированные геометрические параметры и значения энергий взаимодействия показывают, что для подобных систем характерны слабые взаимодействия, что не позволяет адекватно описывать экспериментально наблюдаемые явления и определить наиболее выгодные для диссоциативной адсорбции места на поверхности.
Структуры 2d-2g отражают результаты оптимизации и расчета на уровне теории B3LYP/DGDZVP систем, содержащих четырехатомный кластер серебра, а также молекулярный синглетный кислород в различных положениях. Видно, что среди представленных структур только структура 2d (исходное положение молекулы кислорода «atop») является планарной. В указанной структуре относительное уменьшение длин связей r(Ag(2)-Ag(3)) и r(Ag(3)-Ag(4)) обусловлено смещением зарядовой плотности от кластера серебра к молекуле кислорода. Малликеновские заряды на атомах серебра Ag(1) и Ag(3) составляют 0.097 и 0.274 e. Молекула кислорода расположена отдельно от кластера и находится в его плоскости. Величина малликеновского заряда на каждом атоме составляет -0.265 e. При этом значение длины связи в молекуле кислорода несколько превышает экспериментальную величину. Значение рассчитанной энергии взаимодействия позволяет предполагать наличие слабых взаимодействий в системе.
Из Таблицы 2 можно видеть, что структуры 2e-2g являются трехмерными. Оптимизированная структура 2e, соответствующая положению «bridge» молекулы синглетного кислорода, представляет собой скошенный [38] кластер Ag4, над одной из связей которого располагается молекула кислорода. Необходимо отметить также, что в представленной структуре расстояние между атомами кислорода составляет 1.4 Å.
В оптимизированных структурах 2f и 2g, соответствующих положениям «hollow, above» и «hollow, in-plane», атомы серебра в кластере координируются в структуру, близкую к тетраэдрической. В структуре 2f расстояние между атомами кислорода составляет 1.433 Å. Структура 2g координирована в состояние, в котором один из атомов кислорода располагается между двумя атомами серебра, а второй – близко к центру воображаемой грани тетраэдра. Данная структура может быть использована при трактовке процессов образования приповерхностного кислорода [9-16]. Расстояние между атомами кислорода составляет 1.548 Å.
Наиболее интенсивные полосы в рассчитанных ИК спектрах оптимизированных структур 2d-2g соответствуют диссоциативным колебаниям в молекуле синглетного кислорода. Можно говорить о том, что имеет место тенденция к диссоциации молекулы кислорода и переходу образовавшихся атомов в хемосорбированное состояние. Это предположение также подтверждается увеличением расстояния r(O-O) для всех структур по сравнению с экспериментально определенными значениями для изолированной молекулы.
В Таблице 1 представлены результаты расчетов полных энергий структур 2d-2g, а также энергий взаимодействия молекул синглетного кислорода с кластером серебра. Полученные значения показывают, что наиболее энергетически выгодным является положение молекулы кислорода «hollow, above». Чуть менее выгодным является положение «bridge». Зависимость энергии взаимодействия в системе «кластер серебра – молекулярный синглетный кислород» от расстояния между атомами кислорода в оптимизированной структуре представлена на Рисунке 2. Видно, что зависимость имеет выраженный экстремум в области длин расстояний между атомами кислорода 1.40-1.45 Å.
Необходимо отметить, что в структурах 2d и 2g, обладающих меньшими значениями полной энергии и энергии взаимодействия, расстояния между атомами кислорода и серебра достаточно хорошо коррелируют с известными экспериментальными значениями [37]. При этом расстояния от молекулы кислорода до поверхности серебра (структура 2f) и ребра между двумя соседними атомами серебра (структура 2e) составляют соответственно 1.833 и 1.194 Å. Вероятно, меньшая энергетическая выгодность структуры 2e по сравнению со структурой 2f обусловлена слишком близким расположением кислорода относительно атомов серебра, в то время как для последней этот показатель, по-видимому, соответствует некоторому оптимальному значению.
Таким образом, все представленные структуры позволяют качественно объяснять экспериментальные наблюдения. Уровень теории B3LYP/DGDZVP позволил получить более точные по сравнению с методом HF/DGDZVP данные об энергиях взаимодействия, поскольку учитывает электронную корреляцию. Принимая во внимание результаты определения полных энергий систем можно предположить, что наиболее благоприятным для диссоциативной физической адсорбции кислорода на поверхности серебра является положение «hollow, above».
3.4. Термохимические расчеты
В Таблице 3 приведены результаты термохимических расчетов некоторых из исследованных систем исходя из данных, полученных на уровне теории B3LYP/DGDZVP. Расчеты выполнены для температуры 830 K, которая довольно часто достигается в промышленных условиях реализации окислительных каталитических процессов. Формы Z и ZO2 соответствуют активному центру и молекулярной адсорбированной форме кислорода на поверхности катализатора.
Полученные в настоящей работе значения теплот диссоциативной адсорбции кислорода хорошо согласуются с экспериментальными значениями. Показано [22 и ссылки в ней], что для процесса адсорбции молекулярного кислорода на поверхности грани серебра (110) теплоты молекулярной адсорбции находятся в интервале -4.8 ± -11 ккал/моль, в то время как теплоты диссоциативной адсорбции составляют -40 – -60 ккал/моль. Для случая адсорбции кислорода на грани серебра (111) указанные параметры входят в интервалы -9.2 – -10.6 ккал/моль и -22 – -40.8 ккал/моль, соответственно. Таким образом, рассчитанные термодинамические параметры достаточно хорошо согласуются с данными эксперимента.
Выводы
В рамках настоящей работы проведено детальное исследование механизма адсорбции молекулярного кислорода на поверхности четырехатомного кластера серебра на уровнях теории HF/DGDZVP и B3LYP/DGDZVP. Показано, что DFT методы расчета позволяют с большей точностью оценивать полные энергии систем и энергии адсорбционного взаимодействия при относительно небольших затратах времени и компьютерных ресурсов. Кроме того, геометрические параметры, определенные по результатам DFT расчетов, гораздо лучше коррелируют с экспериментальными значениями. Определены полные энергии кластера серебра Ag4, а также систем «адсорбат-поверхность», моделирующих процессы диссоциативной физической адсорбции молекулярного кислорода. По результатам расчетов установлено, что наиболее выгодным для диссоциативной адсорбции молекулярного кислорода является положение «hollow, above». Подтверждено, что в окислительных каталитических процессах, протекающих на поверхности серебра, с большей вероятностью участвует молекулярный кислород в синглетном состоянии.
Литература
[1]. S. Kagawa, M. Iwamoto, H. Mori, T. Seiyama. J. Phys. Chem. 1981. Vol. 85. p. 434.
[2]. I. E. Wachs. Surf. Sci. 2003. Vol. 544. p. 1.
[3]. O. V. Magaev, A. S. Knyazev, O. V. Vodyankina, N. V. Dorofeeva, A. N. Salanov, A. I. Boronin. Appl. Catal. A. 2008. Vol. 344. p. 142.
[4]. A. Nagy, G. Mestl. Appl. Catal. A. 1999. Vol. 188. p. 337.
[5]. Y. Lei, F. Mehmood, S. Lee, J. Greeley, B. Lee, S. Seifert, R. E. Winans, J. W. Elam, R. J. Meyer, P. C. Redfern, D. Teschner, R. Schlögl, M. J. Pellin, L. A. Curtiss, S. Vajda. Science. 2010. Vol. 328. p. 224.
[6]. W. W. Smeltzer, L. Tollefson, A. Cambron. Canad. J. Chem. 1956. Vol. 34. p. 1046.
[7]. G. I.N. Waterhouse, G. A. Bowmaker, J. B. Metson. Appl. Surf. Sci. 2003. Vol. 214. p. 36.
[8]. V. V. Kaichev, V. I. Bukhtiyarov, M. Hävecker, A. Knop-Gercke, R. W. Mayer, R. Schlögl. Kinet. Catal. 2003. Vol. 44. p. 432.
[9]. C. Rehren, M. Muhler, X. Bao, R. Schlogl, G. Ertl. Z. Phys. Chem. 1991. Vol. 174. p. 11.
[10]. X. Bao, M. Muhler, B. Pettinger, Y. Uchida, G. Lehmpfuhl, R. Schlögl. Catal. Lett. 1995. Vol. 32. p. 171.
[11]. V. I. Bukhtiyarov, A. I. Boronin, M. P. Oschepkova, V. I. Savchenko. React. Kinet. Catal. Lett. 1989. Vol. 39. p. 21.
[12]. C. Rehren, G. Isaac, R. Schlögl, G. Ertl. Catal. Lett. 1991. Vol. 11. p. 253.
[13]. J. Greeley, M. Mavrikakis. J. Phys. Chem. C. 2007. Vol. 111. p. 7992.
[14]. Z. Qu, M. Cheng, W. Huang, X. Bao. J. Catal. 2005. Vol. 229. p. 446.
[15]. G. I.N. Waterhouse, G. A. Bowmaker, J. B. Metson. Appl. Catal. A. 2004. Vol. 265. p. 85.
[16]. C. Stegelmann, P. Stoltze. Surf. Sci. 2004. Vol. 552. p. 260.
[17]. V. A. Sobyanin, V. V. Gorodetskii, N. I. Bulgakov. React. Kinet. Catal. Lett. 1975. Vol. 3. p. 223.
[18]. L. Jia, Y. Wang, K. Fan. J. Phys. Chem. B. 2003. Vol. 107. p. 3813.
[19]. V. I. Avdeev, A. I. Boronin, G. M. Zhidomirov. J. Struct. Chem. 2002. Vol. 43. p. 26.
[20]. V. I. Avdeev, S. F. Ruzankin, G. M. Zhidomirov. J. Struct. Chem. 1997. Vol. 38. p. 519.
[21]. C. Qin, J. L. Whitten. J. Phys. Chem. B. 2005. Vol. 109. p. 8852.
[22]. E. German, I. Efremenko. J. Mol. Struct.: Theochem. 2004. Vol. 711. p. 159.
[23]. R. Poteau, J.-L. Heully, F. Spiegelmann. Z. Phys. D. 1997. Vol. 40. p. 479.
[24]. Z. Tian, Z. Tang. Rapid Commun. Mass Spectrom. 2005. Vol. 19. p. 2893.
[25]. P. Weis, T. Bierweiler, S. Gilb, M. M. Kappes. Chem. Phys. Letters. 2002. Vol. 335. p. 355.
[26]. E. M. Fernandez, M. B. Torres, L. C. Balbas. Recent Adv. Theory Chem. Phys. Syst. 2006. p. 407.
[27]. V. Romanov, C.-K. Siu, U. H. Verkerk, H. El Aribi, A. C. Hopkinson, K. W. Siu. J. Phys. Chem. A. 2008. Vol. 112. p. 10912.
[28]. O. Kh. Poleshchuk, A. G. Yureva, V. D. Filimonov, G. Frenking. J. Mol. Struct.: Theochem. 2009. Vol. 912. p. 67.
[29]. A. Kaczor, K. Malek, M. Baranska. Pyridine on Colloidal Silver. J. Phys. Chem. C. 2010. Vol. 114. p. 3909.
[30]. Gaussian 03, Revision B03. M. J. Frisch, G. W. Trucks, H. B. Schlegel, P. M.W. Gill, B. G. Johnson, M. A. Robb, J. R. Cheeseman, T. Keith, G. A. Petersson, J. A. Montgomery, K. Raghavachari, M. A. Al-Laham, V. Zakrzewski, J. V. Ortiz, J. B. Foresman, J. Closlowski, B. B. Stefanov, A. Nanayakkara, M. Challacombe, C. Y. Peng, P. Y.Ayala, W. Chen, M. W. Wong, J. L. Andress, E. S. Replogle, R. Gomperts, R. L. Martin, D. J. Fox, J. S. Binkley, D. J. Defress, J. Baker, J. P. Stewart, Head-Gordon, C. Gonzales, J. A. Pople. Gaussian, Inc: Pittsburg, PA, 2003.
[31]. J. B. Foresman, E. Frisch. Exploring chemistry with electronic structure methods. second ed., Gaussian Inc., 1996.
[32]. N. W. Ashcroft, N. D. Mermin. Solid State Physics, Holt, Rinehart, and Winston, New York, 1976.
[33]. J. W. Ochterski, Thermochemistry in Gaussian, Gaussian Inc., 2000.
[34]. J. Robinson, D. P. Woodruff. Surf. Sci. 2004. Vol. 556. p. 193.
[35]. R. D. Jones, D. mmerville, F. Basolo. Chem. Rev. 1979. Vol. 79. p. 139.
[36]. E. V. Boikov, M. V. Vishnetskaya, A. N. Emel’yanov, Yu. N. Rufov, N. V. Shcherbakov, I. S. Tomskii. Russ. J. Phys. Chem. A. 2007. Vol. 81. p. 861.
[37]. L. Koponen, L. O. Tunturivuori, M. J. Puska, Y. Hancock. J. Chem. Phys. 2010. Vol. 132. p. 14301.
[38]. J. W.de M. Carneiro, M. T. de M. Cruz. J. Phys. Chem. A. 2008. Vol. 112. p. 8929.
Таблица 1 – Результаты расчетов энергий основного состояния и энергий взаимодействия систем адсорбат/поверхность на уровнях теории HF/DGDZVP и B3LYP/DGDZVP
№№ | Рассчитываемая система | Метод | EAg | E(g) | TE | Einter |
1 | Ag4 + O2(atop) singlet | HF/DGDZVP | -20788,0681482 | -149,5415023 | -20937,6799314 | -0,0702809 |
B3LYP/DGDZVP | -20798,0026051 | -150,2889675 | -20948,3365524 | -0,0449798 | ||
2 | Ag4 + O2(atop) triplet | B3LYP/DGDZVP | -20798,0026051 | -150,3504269 | -20948,3666519 | -0,0136199 |
3 | Ag4 + O2(bridge) singlet | HF/DGDZVP | -20788,0681482 | -149,5415023 | -20937,6799314 | -0,0702809 |
B3LYP/DGDZVP | -20798,0026051 | -150,2889675 | -20948,3538201 | -0,0622475 | ||
4 | Ag4 + O2(bridge) triplet | B3LYP/DGDZVP | -20798,0026051 | -150,3504269 | -20948,3732137 | -0,0201817 |
5 | Ag4 + O2(hollow, above) singlet | HF/DGDZVP | -20788,0681482 | -149,5415023 | -20937,6126432 | -0,0029927 |
B3LYP/DGDZVP | -20798,0026051 | -150,2889675 | -20948,3562707 | -0,0646981 | ||
6 | Ag4 + O2(hollow, above) triplet | B3LYP/DGDZVP | -20798,0026051 | -150,3504269 | -20948,3732136 | -0,0201816 |
7 | Ag4 + O2(hollow, in-plane) singlet | HF/DGDZVP | -20788,0681482 | -149,5415023 | -20937,6799303 | -0,0702798 |
B3LYP/DGDZVP | -20798,0026051 | -150,2889675 | -20948,3386589 | -0,0470863 | ||
8 | Ag4 + O2(hollow, in-plane) triplet | B3LYP/DGDZVP | -20798,0026051 | -150,3504269 | -20948,3731994 | -0,0201674 |
EAg – энергия четырехатомного кластера серебра Ag4, a. u.; E(g) – энергия адсорбированной молекулы в газовой фазе, a. u.; TE – полная энергия системы адсорбат/поверхность, a. u.; Einter – энергия взаимодействия всех компонентов системы, a. u.
Таблица 2 – Оптимизированные на уровнях теории HF/DGDZVP и B3LYP/DGDZVP структуры для процессов физической адсорбции молекулярного кислорода в синглетном и триплетном состояниях на поверхности кластера серебра Ag4.
|
|
| |
A | b | c | |
r(Ag(1)-Ag(2))=3.040, r(Ag(1)-Ag(4))=3.040, r(Ag(2)-O(6))=r(Ag(4)-O(5))=2.251, r(O(6)-Ag(3))=r(Ag(3)-O(5))=2.260. Angle: (O-Ag-O)= 38.3, (Ag(2)-Ag(1)-Ag(4))= 58.1, (Ag(1)-Ag(2)-Ag(3))=131.9. | r(Ag(1)-Ag(2))=r(Ag(2)-Ag(3))=3.104, r(Ag(3)-Ag(4))=r(Ag(4)-Ag(1))=3.087, r(Ag(2)-O(5))=2.940. Angle: (Ag(1)-Ag(2)-Ag(3))=124.9, (Ag(3)-Ag(4)-Ag(1))=126.1, (Ag(2)-Ag(1)-Ag(4))=(Ag(2)-Ag(3)-Ag(4))=54.5. | Triplet: r(Ag(2)-Ag(3))=3.151, r(Ag(1)-Ag(4))=2.919, r(Ag(1)-Ag(2))=2.826, r(Ag(4)-Ag(3))=2.846, r(O-O)=1.325. Angle: (Ag(1)-Ag(2)-Ag(3))=60.1, (Ag(1)-Ag(4)-Ag(3))=57.1, (Ag(4)-Ag(3)-Ag(2))=127.2, (Ag(4)-Ag(1)-Ag(2))=117.7. | |
|
|
|
|
d | e | f | g |
Singlet: r(Ag(1)-Ag(2))=r(Ag(1)-Ag(4))=2.886, r(Ag(2)-Ag(3))=r(Ag(3)-Ag(4))=2.840, r(O-O)=1.339. Angle: (Ag(4)-Ag(1)-Ag(2))=120.3, (Ag(2)-Ag(3)-Ag(4))=123.7. Triplet: r(Ag(1)-Ag(2))=r(Ag(1)-Ag(4))=2.871 , r(Ag(2)-Ag(3))=r(Ag(3)-Ag(4))=2.914, r(O-O)=1.285 Angle: (Ag(4)-Ag(1)-Ag(2))=124.4o , (Ag(1)-Ag(2)-Ag(3))=57.2o | r(Ag(1)-Ag(2))=2.842, r(Ag(1)-Ag(4))=2.834, r(Ag(3)-Ag(4))=3.071, r(Ag(2)-Ag(3))=4.498 Å,r(O-O)=1.4. Angle: (Ag(1)-Ag(4)-Ag(3))=140.1, (Ag(2)-Ag(1)-Ag(4))= 61.6 , (Ag(2)-Ag(3)-Ag(4))=39.8 | r(Ag(1)-Ag(2))=2.914, r(Ag(1)-Ag(3))=3.259, r(Ag(1)-Ag(4))=3.197, r(Ag(3)-Ag(4))=2.975, r(Ag(2)-Ag(3))=2.957, r(Ag(2)-Ag(4))=2.931, r(O-O)=1.433. Angle: (Ag(3)-Ag(1)-Ag(4))=54.9, (Ag(1)-Ag(4)-Ag(3))=63.6, (Ag(4)-Ag(3)-Ag(1))=61.5. | r(Ag(2)-Ag(3))=2.892, r(Ag(2)-Ag(4))=2.884, r(Ag(3)-Ag(4))=2.868, r(Ag(2)-O(5))=2.348, r(Ag(1)-O(5))=2.239, r(O-O)=1.548. Angle: (Ag(2)-Ag(3)-Ag(4))=60.1, (Ag(3)-Ag(4)-Ag(2))=60.4, (Ag(4)-Ag(2)-Ag(3))=59.5. |
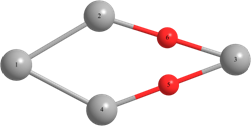
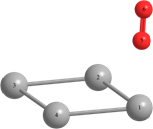
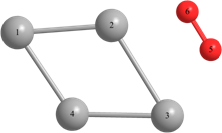
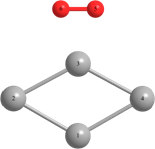
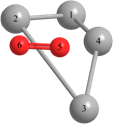
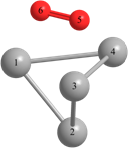
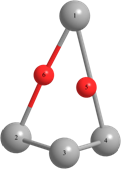
Длины связей (r) выражены в Å, углы между атомами выражены в градусах (o).
Таблица 3 – Результаты термохимических расчетов процесса диссоциативной адсорбции молекулы кислорода в синглетном состоянии на кластере серебра Ag4 на уровне теории B3LYP/DGDZVP.
Реакция | Вариант реакции | ΔH (830), ккал/моль | ΔG (830), ккал/моль |
| O2 + Z = ZO2 (atop) | -26,14 | -3,70 |
O2 + Z = ZO2 (bridge) | -37,07 | -11,62 | |
O2 + Z = ZO2 (hollow, above) | -38,66 | -13,03 | |
O2 + Z = ZO2 (hollow, in-plane) | -27,65 | -2,14 |
Рисунки
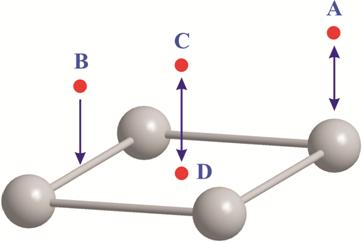
Рисунок 1 – Общий вид исходных систем.
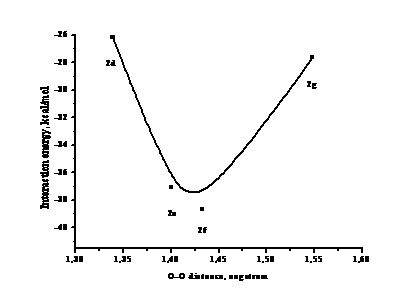
Рисунок 2 – Зависимость энергии взаимодействия от расстояния между атомами кислорода
