

Партнерка на США и Канаду по недвижимости, выплаты в крипто
- 30% recurring commission
- Выплаты в USDT
- Вывод каждую неделю
- Комиссия до 5 лет за каждого referral
УДК 541.64:539.(199+3)
Влияние заместителей при двойной С=С связи на механизм присоединения озона
© ,1 ,2*
,1 1
и 1
1 Лаборатория физико-химии композиций синтетических и природных полимеров. Институт Биохимической Физики им. ,. ИБХФ РАН, Ул. Косыгина, 4.
г. Москва, 119334. Россия. Тел.: (495) 939-71-93. E-mail: fizhim@rambler.ru…
2 Лаборатория кинетики гетерофазных процессов. Институт проблем химической физики, ИПХФ РАН. пр-т Академика Семенова, 1. г. Черноголовка, 142432. Московская область. Россия.
Тел.: (495) 993-57-07 E-mail: bkris@mail.ru
Ключевые слова: озон, этилен, бутен-2, этилен хлорзамещенный, кинетика реакций
Аннотация
В работе представлены результаты расчета первичной стадии реакции озона с двойной связью этилена, бутена-2, хлорпроизводными этилена (от C2Cl4 до C2H3Cl). Расчет выполняли аb initio методом MRMP2 и методом DFT (B3LYP) с использованием базисов 6-31+G/6-311+G с диффузными функциями и aug-cc-PVDZ. Изучены кинетические закономерности двух путей реакции – согласованное присоединение через симметричное переходное состояние (механизм Криге) и несогласованное присоединение через бирадикальное переходное состояние (маханизм ДеМура). Оба эти механизма удалось удачно описать как в однодетерминантным так и многодетерминантным приближении. Расчеты подтвердили наличие обоих каналов для всех реагентов и дали разумные величины для констант скорости, согласующиеся с эксперементом.
Введение
Взаимодействие озона с двойной связью – это одна из наиболее специфических реакций непредельных соединений. Поэтому механизм этой реакции интенсивно изучали как теоретически, так и экспериментально последние лет 60 [1-3]. До недавнего времени считалось, что реакция протекает по механизму 1,3-циклоприсоединения через симметричное переходное состояние (ПС1) с образованием в первом акте пятичленного циклического молозонида или первичного озонида (механизм Криге [4]):
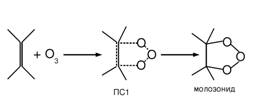
Эта схема реакции подтверждена многочисленными прямыми и косвенными экспериментальными данными [1, 2].
Альтернативный механизм реакции, при котором озон реагирует с двойной связью подобно пероксильному радикалу с образованием промежуточного бирадикального переходного состояния ПС2, был предложен ДеМуром в [5] для реакции озона с ацетиленом в попытке объяснения ее аррениусовских параметров:
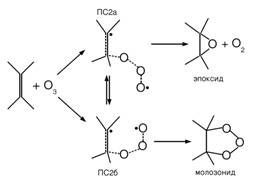
В первых работах группы [6-8], посвященных реакции присоединения озона к двойной связи объектом исследований был этилен. Вопрос заключался в выяснении возможных механизмов данной реакции и их вклада в общую константу скорости реакции. Результаты квантово-химических расчетов показали, что действительно наряду с принятым в литературе симметричным переходным состоянием (ПС1) [9-11] существует несеметричное переходное состояние (ПС2) [7] вклад которого для данной реакции минимален. В дальнейших работах [13-18] преследовалось две цели. Во первых, необходимо было установить влияние заместителей при двойной связи на конкуренцию механизмов согласованного (через ПС1) и несогласованного (через ПС2) присоединения, а во вторых, отобрать метод или комбинацию методов, способных дать в широком диапазоне реагентов разумные значения кинетических параметров для исследуемых реакций. Расчеты показали, что для олефинов приемущественным каналом является механизм согласованного присоединения, в то время как для фтор и хлор замещенных этилена в некоторых случаях реакция идет преимущественно через ПС2. Результаты квантово-химических расчетов были подтверждены полуэмпирическим методом пересекающихся парабол [19].
Было установлено [18], что оптимальными методами для описания соотношения каналов и кинетики реакций олефинов и хлорзамещенных этилена с озоном служат ab initio метод MRMP2 и DFT метод B3LYP. В настоящей работе данными методами исследовали первичную стадию озонолиза этилена, бутена-2 и хлорпроизводных этилена – от C2Cl4 до C2H3Cl.
Методика расчета
Расчеты выполняли в вычислительном центре ИПХФ РАН (Черноголовка) с помощью программы GAUSSIAN-03 [20] и US GAMESS ver.7 [21] и PC GAMESS ver 7.0 [22] как для закрытых, так и для открытых оболочек (для ограниченного и неограниченного методов Хартри-Фока или Кона-Шама). Использовали наборы базисных функций семейства 6-31G/6-311G c диффузными функциями и aug-cc-PVDZ. Параметры состояний, соответствующих минимумам на потенциальной поверхности, находили при полной оптимизации всех переменных, а переходное состояние - используя геометрию состояний, находящихся до и после ПС на координате реакции. В экстремальных точках потенциальной поверхности вычисляли частоты нормальных колебаний. Многоконфигурационный расчет MRMP2 выполняли используя геометрию соответствующего состояния, полученную на уровне CASSCF [18]. Используемые методы проверяли на размерную-несогласованность, для этого выполняли сканирование координаты реакции в обе стороны от переходного состояния. При выходе энергии на плато в области больших значений расстояния между реагирующими атомами (2,5 – 4,0 Å), сканирование прекращали и полученное значение энергии брали в качестве энергии реагентов. Размер активного пространства для этилена, бутена-2, хлорпроизводных этилена брали (2, 2), то есть два электрона на двух орбиталях (связывающая и разрыхляющая π-орбитали). Для озона активное пространство варьировали от (6, 6) до (12, 9). Следует отметить, что в литературе применяли для озона различные активные пространства, однако, по-видимому, наиболее разумные обоснования для его величины приведены в работах [23 – 25], где обосновывается активное пространство величиной (8, 7) и (12, 9) для озона, что соответствует размерам (10, 9) и (14, 11) для комплексов и переходных состояний.
Значения энтальпии и энтропии реакции вычисляли с использованием результатов квантовых расчетов с помощью программы MOLTRAN [26]. Значения констант скорости вычисляли по стандартной теории переходного состояния с использованием результатов квантовых расчетов и расчетов термодинамических величин.
Результаты и их обсуждение
Методами B3LYP и MRMP2 для всех исследуемых в работе реакций получали переходные состояния по механизму согласованного и несогласованного присоединения. Для ПС1 характерно практически равные расстояния С…О (Rco) в каждой паре атомов и нулевое значение S2 в случае однодетерминантного приближения, что неоднократно анализировалось ранее [6]. Тогда как для ПС2 характерны разные длины C…O (Rco), молекула озона развернута относительно двойной связи, а значение S2≈0.7.
Результаты сканирования координаты реакции от ПС в сторону реагентов, то есть в сторону увеличения расстояния между реагирующими атомами С и О, показали, что энергия реагентов, находящихся на расстоянии 3-4А, в задачах расчитанных методом B3LYP незначительно ниже (на 1-3 кДж/моль), а для задач, посчитанных методом MRMP2, значительно ниже (на 50-70 кДж/моль) суммарной энергии исходных веществ, посчитанных теми же методами. Учет данной размерной несогласованности позволяет избежать существенной переоценки константы скорости, которую традиционно получали без сканирования координаты реакции исходя из энергий ПС и суммы энергий исходных веществ.
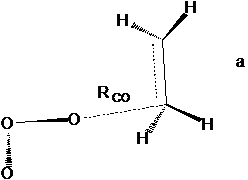
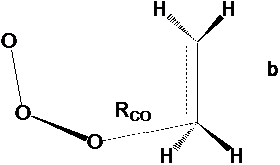
Рис. 1 Структура переходного состояния реакции этилена с озоном по механизму несогласованного присоединения: (а) – транс-ПС2; (б) – цис-ПС2 .
В таблице 1. представлены результаты расчетов энергий ПС, а также констант скорости для реакции этилена с озоном. Согласно литературным данным константа скорости озона с этиленом в газовой фазе составляет k = 103 л*(моль*с)-1 [27], энергия активации около 20 кДж/моль. В реакции по механизму ДеМура (рис.1) для данной реакции возможны две конфигурации ПС, отличющиеся расположением озонового фрагмента относительно двойной связи – транс-конфигурация и цис-конфигурация, причем цис-конфигурация более выгодна. Следует отметить, что эти конфигурации достаточно близки по энергии и все выводы для них практически идентичны. В случае наличия обоих конфигураций расчет выполняли для более выгодного состояния.
Из таблицы видно что метод B3LYP достаточно неплохо описывает величины энергий активации и значения констант скорости для механизма согласованного и несогласованного присоединения, однако значения энергии ПС1 получаются несколько заниженными, что приводит к завышенному значению константы скорости по механизму Криге (k1). Соотношение каналов реакции получается разумным при всех базасих (k1/k2 ~ 102, k2 – скорость реакции по механизму ДеМура). Результаты MRMP2 (табл.1) расчета слабо зависят от размера активного пространства, во всех случаях энергии активации для механизма Криге получаются около 20 кДж/моль, что согласуется с экспериментом, а для механизма ДеМура – около 50. Отношение скоростей при реакции по двум каналам в этих расчетах составляет 104 – 105 в пользу механизма Криге, что также согласуется с литературными данными. В целом, результаты многоконфигурационного расчета (MRMP2) согласуются с данными для однодетерминантного приближения (B3LYP).
Метод | Базис | Ea, kJ/mol | k∙10-3, л*моль-1с-1 | |||
ПС1 | ПС2 | k1 | k2 | k1/ k2 | ||
B3LYP | 6-31+G** | 2,9 | 22,8 | 37,6 | 3,1E-01 | 1,2E+02 |
aug-cc-PVDZ | 5,9 | 27,17 | 2,94 | 1,6E-02 | 1,8E+02 | |
MRMP2(10_9) | 6-31+G** | 25,7 | 58,8 | 2,13 | 1,2E-04 | 1,0E+04 |
MRMP2(10_10) | 6-31+G** | 20,3 | 58,9 | 21,14 | 6,3E-05 | 1,0E+05 |
MRMP2(14_11) | 6-31+G** | 24,8 | 54,1 | 1,7 | 5,1E-04 | 1,0E+05 |
Табл. 1 Параметры переходных состояний по данным ab initio и DFT расчетов: энергия активации Еa (кДж/моль), константы скорости реакции по механизму Криге k1 и Демура k2 (л*(моль*с) -1, 298 К) и их отношение.
В реакции бутена-2 с озоном по механизму ДеМура, как и для этилена, возможно образование ПС двух типов, отличающихся расположением озонового фрагмента относительно двойной связи: цис-ПС2 и транс-ПС2. В случае реакции цис-бутена-2 с озоном по механизму Криге, из-за отсутствия осевой симметрии возможны два варианта ПС, которые отличаются друг от друга положением озона относительно плоскости молекулы бутена-2. В первом случае молекула озона наклонена в сторону СН3-групп во втором от них. Анализ реакционной способности выполняли для тех ПС, энергия которых ниже, т. е. реакция через них протекает быстрее. В случае ПС2 выбор переходного состояния практически не влияет на результаты расчетов кинетики реакции.
В таблице 2 представлены результаты расчетов энергий ПС а также констант скорости для реакции бутена-2 с озоном. Согласно литературным данным константа скорости озона с бутеном-2 в газовой фазе при нормальных условиях по порядку величины равна 105 л*(моль*с)-1, энергия активации этой реакции составляет 9 – 10 кДж/моль [28]. При этом реакции цис- и транс-бутена-2 с озоном с точки зрения кинетики отличаются незначительно [28]. Результаты расчетов как методом B3LYP, так и MRMP2 (табл. 2) в целом соответствуют экспериментальным данным. Расчеты B3LYP при этом дают разумную величину для константы скорости реакции по механизму Криге, но значительно завышают скорость реакции по механизму ДеМура, тогда как согласно всем предварительным оценкам (см. выше) в олефинах должен преобладать первый канал реакции. Данные многоконфигурационных расчетов позволяют заключить, что для данной задачи активное пространство (10,9) недостаточно, гораздо лучше соответствуют эксперименту данные с пространством размером (14,11). Обращает на себя внимание, что расчеты в таком активном пространстве дают разумные результаты, но при использовании базисного набора 6-31+G** значение энергии активации получается несколько завышенное. Мы предположили, что это следствие малости данного базиса и выполнили расчет с использованием набора aug-cc-pVDZ. Действительно, в этом случае значение энергии активации получается ниже, оно соответствует экспериментальным данным, как и само значение константы скорости. Соотношение каналов реакции тоже не противоречит предварительным оценкам.
Вещество | Метод | Базис | Ea, kJ/mol | k, л*моль-1с-1 | |||
ПС1 | ПС2 | k1 | k2 | k1/ k2 | |||
цис-бутен-2 | B3LYP | 6-31+G** | 1,15 | 5,95 | 1,42E+05 | 3,26E+05 | 4,35E-01 |
B3LYP | 6-311+G** | 2,13 | 8,38 | 5,45E+04 | 1,10E+05 | 4,95E-01 | |
MRMP2(10_9) | 6-31+g** | 14,30 | 42,64 | 4,67E+01 | 7,14E-04 | 6,54E+04 | |
MRMP2(14_11) | 6-31+g** | 18,18 | 36,51 | 3,84E+04 | 7,45E+01 | 5,16E+02 | |
MRMP2(14_11) | aug-cc-pvdz | 10,06 | 26,51 | 1,02E+06 | 4,21E+03 | 2,42E+02 | |
транс-бутен-2 | B3LYP | 6-31+g** | -4,26 | 4,63 | 4,76E+05 | 2,26E+05 | 2,11E+00 |
B3LYP | 6-311+G** | -3,37 | 6,72 | 6,50E+05 | 1,93E+05 | 3,36E+00 | |
MRMP2(10_9) | 6-31+g** | 17,92 | 40,94 | 2,91E+00 | 1,01E-03 | 2,87E+03 | |
MRMP2(14_11) | 6-31+g** | 21,62 | 34,85 | 1,10E+04 | 5,05E+00 | 2,18E+03 | |
MRMP2(14_11) | aug-cc-pvdz | 14,42 | 28,71 | 2,01E+05 | 6,01E+01 | 3,35E+03 |
Табл. 2 Параметры переходных состояний по данным ab initio и DFT расчетов (обозначения как в табл. 1).
Рассмотрим реакцию хлорзамещенных этилена с озоном. Для симметричных хлорзамещенных этилена (тетрахлорэтилен, транс-СHCl-CHCl и СCl2-CH2) возможно образование только одного ПС1 так же как для этилена. В остальных случаях возникают два ПС1 – когда центральный атом кислорода в озоне наклонен в сторону, где больше атомов хлора (ПС1а) и в противоположную сторону (ПС1б) – см. рис.2.1. Конфигурации ПС2 рассмотрим на примере трихлорэтилена. Здесь в реакции по механизму Демура возможны два варианта ПС – молекула озона находится в положениях цис- и транс- по отношению к двойной связи – цис-ПС2 и транс-ПС2, соответственно (рис. 2.2). При атаке озона на атом углерода, при котором находится один атом хлора, в обоих положениях возможны состояния, при которых центральный атом кислорода молекулы озона “наклонен” в сторону хлора и от него – цис-ПС2а и цис-ПС2б. Кроме того, атака озона возможна на атом углерода, при котором находится различное количество атомов хлора, обозначаемое числом в скобках – цис-ПС2б(1) и цис-ПС2б(2).
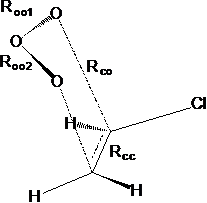
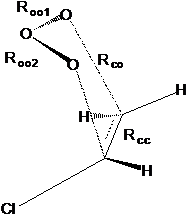
Рис.2.1. Структура переходных состояний реакции по механизму Криге - ПС1а и ПС1б.
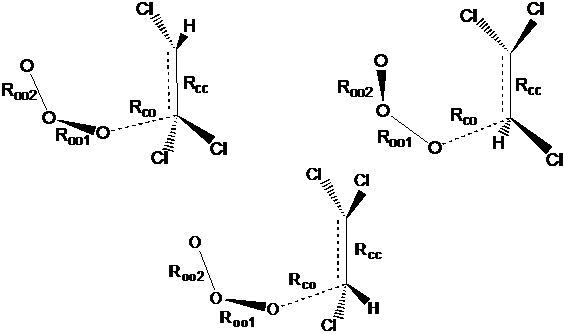 |
Рис.2.2. Структура переходных цис-состояний реакции по механизму Демура: цис-ПС2а(1), цис-ПС2б(1), цис-ПС2а(2) .
Анализ ПС по механизму Криге показал, что в случае снятия симметрии заместителей (C2HCl3, cis-СHCl–CHCl, C2H3Cl) наиболее выгодным всегда является ПС1 с “ориентацией” среднего кислорода к большинству атомов водорода. Различие между ПС1а и ПС1б возрастает с увеличением числа атомов хлора и различия в их количестве с одной и другой стороны молекулы этилена. Так, для C2H3Cl отношение констант скорости реакций через ПС1а и ПС1б составляет 1.24, тогда как для cis-(СHCl)2 – 20, для C2HCl3 – 10.Анализ ПС по механизму Демура показал, что наиболее выгодна атака атома углерода, при котором наибольшее число атомов водорода, т. е. отношение констант скорости реакции достигает нескольких порядков: для C2HCl3 это 103, для CCl2–CH2 – 108, для C2H3Cl – 104. Для симметричной молекулы С2Cl4 более выгодным является реакция через цис-ПС2.
Вещество | Метод | Базис | k1/k2 | k, л*моль-1с-1 | |
расчет | Эксперимент | ||||
C2Cl4 | UB3LYP | aug-cc-pvdz | - | 0,0359 | 0,6 |
MRMP2(8,8) | 6-31+G** | - | 10,1 | ||
C2H3Cl | UB3LYP | aug-cc-pvdz | 0,003 | 28,4 | 30,1 |
MRMP2(8,8) | 6-31+G** | - | 904 | ||
цис-Н2С2Cl2 | UB3LYP | aug-cc-pvdz | 1,68 | 9,78 | 21,6 |
MRMP2(8,8) | 6-31+G** | 179 | 7,2 | ||
транс-2Н2СCl2 | UB3LYP | aug-cc-pvdz | 4 | 63 | 107 |
MRMP2(8,8) | 6-31+G** | 1000 | 800 | ||
СН2-СCl2 | UB3LYP | aug-cc-pvdz | 0,00047 | 2640 | 4,89 |
MRMP2(8,8) | 6-31+G** | - | 170 | ||
С2Н3СCl | UB3LYP | aug-cc-pvdz | 0,67 | 972 | 148 |
MRMP2(8,8) | 6-31+G** | - | 264,54 | ||
MRMP2(14,11) | 6-31+G** | 0,4 | 1,12 | ||
UB3LYP | 6-31+G** | 0,48 | 1426 |
Табл. 3 Сравнение результатов расчета по методам UB3LYP и MRMP2 соответствующих базисах с экспериментальными данными, k1/k2 – отношение констант скоростей по механизмам Криге и Демура.
В таблице 3 можно заметить, что если отбросить 1,1-дихлорэтилен, то экспериментальные [28] и расчитанные методом B3LYP данные образуют по константам скорости один и тот же ряд: наименьшее значение у C2Cl4, затем — cis-1,2-, затем – С2HCl3, затем trans-1,2-, затем монохлор- и, наконец, этилен. Суммарную константу скорости по обоим каналам вычисляли по формуле k=2k1+nk2, где n — стерический фактор (для механизма Криге n = 2, а для механизма Демура в случаях C2Cl4, cis-(СHCl)2, trans-(СHCl)2, C2H4 этот параметр равен 4, для остальных хлорпроизводных этилена он тоже равен 2). Это еще раз свидетельствует о хорошем описании методом B3LYP таких сложных (по сути многоцентровых) систем и дает основание констатировать, что он полностью неприменим к 1,1-дихлорэтилену. Чтобы выяснить причины этого, мы выполнили дополнительные расчеты методом MRMP2. Прежде всего отметим, что существование обоих ПС удалось подтвердить не только в одно-, но и в многодетерминантном приближении. Так, в MRMP2-расчетах для монохлорэтилена получены оба переходных состояния, но в этом случае не слишком удачно описывается ПС1, для которого существенно занижена энергия активации. Это приводит к завышению константы скорости реакции по механизму Криге и к неверной оценке соотношения каналов реакции.
Таким образом, проверить результаты расчетов B3LYP для каждого из каналов, не используя другие методы или активные пространства не представляется возможным.
Заключение
Данные расчетов показали возможность моделирования реакций присоединения озона к кратным связям, однако всегда необходим всесторонний анализ полученных результатов для того, чтобы определить, каким из них (результатов) можно верить, а какие следует отбросить.
Выводы
1. Первичная стадия реакции озона с кратными связями протекает по двум конкурирующим каналам: по механизму согласованного и несогласованного присоединения.
2. Эффективность каждого из этих каналов реакции зависит от окружения двойной связи: в алкенах преобладает первый канал реакции, тогда как при появлении электроотрицательных заместителей (Cl) все большую роль начинает играть второй канал.
3. На одном уровне расчета в однодетерминантном приближении оба эти канала удается описать DFT методом B3LYP, но в отдельных случаях его результаты неоднозначны.
4. В многодетерминантном приближении оба канала описываются достаточно адекватно, но при этом необходимо использовать значительный объем активного пространства (не менее (14, 11)) и достаточно большой набор базисных функций.
Литература
[1] , Озон и его реакции с органическими соединениями. М.: Наука, 1974.
[2] , // Успехи химии. 1980. Т. 49. № 12. С.2344 – 2376.
[3] , , Физическая химия озона: Из-во МГУ. 1998. 480 С.
[4] Criegee R. // Angew. Chem. 1975. V. 87. P. 765.
[5] DeMore W. B. // Int. J. Chem. Kinetics. 1969. V. 1.№ 1. P. 209.
[6] , , // Хим. физика. 2003. Т. 22. № 9. С. 3.
[7] // Хим. физика. 2006. Т. 25. № 6. С. 13.
[8] , , // Хим. физика. 2007, Т.26, № 6, С.16.
[9] McKee M. L., Rohfing C. M. // Ibid. 1989. V. 111.
[10] Gillies C. W., Gillies J. Z., Suenram R. D. et al. // Ibid. 1991. V. 113. № 7. P. 2412.
[11] Chan W. T., Hamilton I. P. // J. Chem. Phys. 2003. V. 118. № 4. P. 1688.
[12] Wheeler S. E., Ess D. H., Houk K. N. // J. Phys. Chem. A 2008, V. 112, P. 1798-1807
[13] // ЖФХ. 2004. Т. 78. № 4. С. 1.
[14] , , // Хим. физика. 2007, Т.26, № 8, С.22.
[15] , , // Хим. физика. 2008, Т.27, № 9, С.1.
[16] , , // Хим. физика. 2008, Т.27, № 2, С.62.
[17] , , // Химическая физика. 2010. Т. 29. № 9. С. 20.
[18] , // Хим. Физика, 2011, Т.30, в печати
[19] , // Хим. физика. 2007. Т. 26. № 5. С. 34.
[20] Frisch M. J., Trucks G. W., Schlegel H. B., Scuseria G. E., Robb M. A., Cheeseman J. R., Montgomery J. A., Jr., Vreven T., Kudin K. N., Burant J. C., Millam J. M., Iyengar S. S., Tomasi J., Barone V., Mennucci B., Cossi M., Scalmani G., Rega N., Petersson G. A., Nakatsuji H., Hada M., Ehara M., Toyota K., Fukuda R., Hasegawa J., Ishida M., Nakajima T., Honda Y., Kitao O., Nakai H., Klene M., Li X., Knox J. E., Hratchian H. P., Cross J. B., Adamo C., Jaramillo J., Gomperts R., Stratmann R. E., Yazyev O., Austin A. J., Cammi R., Pomelli C., Ochterski J. W., Ayala P. Y., Morokuma K., Voth G. A., Salvador P., Dannenberg J. J., Zakrzewski V. G., Dapprich S., Daniels A. D., Strain M. C., Farkas O., Malick D. K., Rabuck A. D., Raghavachari K., Foresman J. B., Ortiz J. V., Cui Q., Baboul A. G., Clifford S., Cioslowski J., Stefanov B. B., Liu G., Liashenko A., Piskorz P., Komaromi I., Martin R. L., Fox D. J., Keith T., Al-Laham M. A., Peng C. Y., Nanayakkara A., Challacombe M., Gill P. M. W., Johnson B., Chen W., Wong M. W., Gonzalez C., and Pople J. A., Gaussian 03, Revision C.02, Gaussian, Inc., Wallingford CT, 2004.
[21] http://www. msg. ameslab. gov/GAMESS/
[22] Granovsky A. A., PC GAMESS version 7.0, http://classic. chem. /gran/gamess/index. html
[23] Anglada J.M., Bofill J. M. // Chem. Phys. Lett. 1995. V. 243. P. 151.
[24] Anglada J.M., Crehuet R., Bofill J. M. // Chem.-Eur. J. 1999. V. 5. P. 1809.
[25] Ljubic I. , Sabljic A. // J. Phys. Chem. 2002. V. 106. P. 4745.
[26] Ignatov S. K. http://ichem. unn. runnet. ru/tcg/Moltran. htm
[27] DeMore W. B. Sander S. P., Golden D. M., Hampson R. F., Kurylo M. J., Howard C. Y., Ravinshankara A. R., Kolb C. F. , Molina M. J. // JPL Publication 1997. P. 97.
[28] http:// http://kinetics. nist. gov/kinetics/index. jsp
