
Партнерка на США и Канаду по недвижимости, выплаты в крипто
- 30% recurring commission
- Выплаты в USDT
- Вывод каждую неделю
- Комиссия до 5 лет за каждого referral
1.
Определение содержания вещества в пробе люминесцентным методом основано на сравнении интенсивности люминесценции пробы и стандартных образцов. Последние должны отвечать ряду требований:
- содержание определяемого вещества в стандартном образце должно быть точно известно;
- химический состав матрицы (основы) стандартного образца должен быть идентичен (или подобен в практически достижимой мере) матрице пробы;
- стандартный образец и проба должны обладать близкими физическими свойствами.
Метод добавок. Наиболее простой способ приготовления стандартных образцов, соответствующих пробе по составу матрицы, заключается в использовании метода добавок. Его целесообразно использовать в тех случаях, когда состав матрицы пробы неизвестен или меняется от пробы к пробе.
Сущность метода заключается в следующем. Берут три одинаковых образца пробы. Ко второму и третьему образцам добавляют точное количество определяемого вещества. Размеры добавок подбираются с таким расчетом, чтобы содержание определяемого вещества во всех трех образцах пробы после указанной процедуры отвечало соотношению
Cх : (Cх +Dc1 ): (Cх +Dс2 ) = 1:2:3,
где Dc1 и Dс2 - изменение содержания определяемого вещества, вызванное процедурой добавки.
После измерения интенсивности люминесценции всех трех образцов строят график в координатах «I –D(c)». Содержание определяемого вещества в пробе Cх находят путем экстраполяции графика на значение I =0.
Метод градуировочного графика (ограничивающих растворов). В этом методе измеряют интенсивность люминесценции серии стандартных образцов (обычно не менее пяти), охватывающих весь диапазон ожидаемых содержаний определяемого вещества в пробе, и строят график зависимости интенсивности люминесценции от массовой доли определяемого вещества. В идеальном случае градуировочный график должен быть линейным и проходить через начало координат. На практике он оказывается линейным лишь в узком диапазоне содержаний определяемого вещества и редко выходит из начала координат.
2.
Потенциометрическое титрование. Способы нахождения конечной точки титрования.
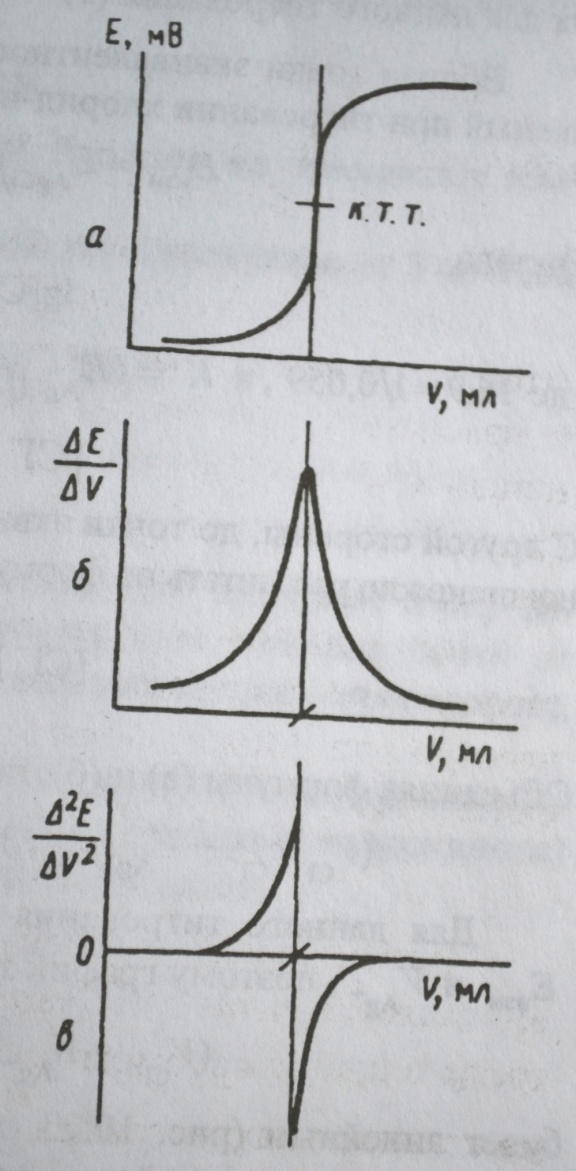
Для нахождения точки эквивалентности кривые потенциометрического титрования строят в различных координатах:
- интегральные кривые титрования в координатах Е(ордината) – V(абсцисса) ;
- дифференциальные кривые титрования в координатах ∆E/∆V – V и ∆2E/∆V2 – V;
где Е – ЭДС потенциометрической ячейки; ∆V - объем добавленного титранта; V – суммарный объем добавленного титранта; ∆E - изменение потенциала, соответствующее добавлению ∆V раствора титранта.
По построенным кривым определяют суммарный объем добавленного титранта V(ТЭ) в точке эквивалентности, и затем рассчитывают количество или концентрацию определяемого вещества.
Как выглядят графики зависимостей в координатах Е – V, ∆E/∆V – V, ∆2E/∆V2 – V.
Как по этим графикам находят объем добавленного титранта V(ТЭ) в точке эквивалентности.
3.
Регенерация катионитов.
При регенерации катионитов колонку наполняют 2 моль/л HCl. Скорость протекания жидкости через ионит обычно 1 мл/мин. Регенерация катионита заканчивается, когда концентрация кислоты в вытекающем из катионита растворе будет равна концентрации исходного раствора.
После пропускания раствора кислоты катионит промывают дистиллированной водой. Полноту отмывания катионита от кислоты проверяют по индикатору метиловому оранжевому.
Катионит, переведенный таким образом в Н+-форму, подготовлен для проведения хроматографического разделения.
4.
Уравнении Ильковича, которое связывает диффузионный ток Id с концентрацией иона с и рядом других величин:
Id = 605 z D1/2 m 2/3 t1/6 c (1)
Где z - заряд иона; D – коэффициент диффузии; m – масса ртути, вытекающей из капилляра за 1 с, мг; t – время образования капли (периода капания), с.
В практике количественного полярографического анализа коэффициент пропорциональности межу концентрацией вещества и силой диффузионного тока обычно устанавливают с помощью стандартных растворов. При постоянных условиях полярографирования D, m, и t постоянны, поэтому уравнение (1) переходит в
Id = k c
5.
Спектр поглощения пробы показывает, на каких длинах волн она преимущественно поглощает излучение внешнего источника. Такие спектры обычно регистрируют в координатах A – л, где А - количественная характеристика поглощения света на данной длине волны, называемая оптической плотностью.
Изучение молекулярных спектров – это важнейший способ количественного химического анализа. Заметим, что количественное определение какого-либо вещества по известной методике вовсе не требует регистрации полного спектра излучения (или поглощения) пробы. Достаточно было бы измерить аналитический сигнал на заранее выбранной длине волны. Спектры нужны для решения гораздо более сложных задач. А именно:
- По спектру индивидуального вещества выбирают ту длину волны, на которой в дальнейшем, в ходе количественного анализа, будут измерять аналитический сигнал этого вещества (I или А). Если для определения какого-либо элемента в атомно-эмиссионном спектральном анализе используют наиболее интенсивные линии эталонного спектра, то в молекулярно-абсорбционном (спектрофотометрическом) анализе аналитический сигнал обычно измеряют на длине волны, соответствующей максимуму на спектральной кривой. Сопоставляя спектры предполагаемых компонентов пробы, выясняют возможность определения одних веществ в присутствии других. Если спектры компонентов пробы накладываются друг на друга, результаты анализа смеси будут завышенными. Для снижения систематических погрешностей, связанных с наложением спектров, созданы особые приемы измерений и расчета результатов. Другие выходы из положения - маскирование или предварительное отделение мешающих компонентов.
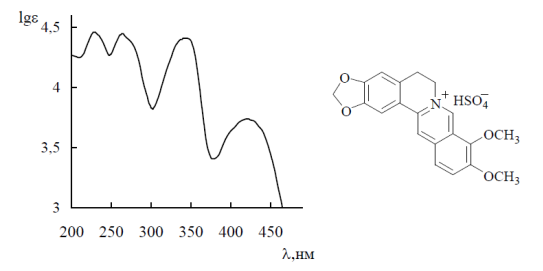
Спектр поглощения алкалоида берберина бисульфата (водный раствор).
6.
Пламенно-ионизационный детектор
Пламенно-ионизационный детектор (ПИД) обладает высокой чувствительностью к органическим соединениям, имеет широкий линейный диапазон и сравнительно малую зависимость рабочих параметров от конструкции и внешних условий.
Для работы детектора необходимо использовать дополнительные газы: воздух и водород. Они поддерживают горение горение пламени в детекторе. Устойчивость работы и максимальная чувствительность ПИД обеспечиваются правильным выбором расходов газа-носителя, водорода и воздуха. Оптимальные расходы газов и их соотношения несколько зависят от конструкции детекторов, однако для большинства конструкций наибольшая чувствительность и стабильность работы достигаются при соотношении расходов газа-носителя, водорода и воздуха, близком к 1:1:10.
Поскольку в пламени чистого водорода число ионов мало, сопротивление газового пространства очень велико и ток детектора весьма мал. Этот ток, возникающий за счет ионизации примесей, содержащихся в газе-носителе, водороде и воздухе, является постоянным фоновым током детектора. При внесении с газом-носителем из колонки анализируемых органических веществ число ионов в пламени резко увеличивается, сопротивление пламени падает и в детекторе регистрируется соответствующее возрастание ионного тока.
Очень слабая реакция ПИД на воду и отсутствие чувствительности к неорганическим соединениям, инертным газам и водороду делают его незаменимым при анализах примесей органических веществ в воздухе, сточных и природных водах, а также в биологических объектах.
Сравнительно слабая зависимость чувствительности детектора от изменения расходов газов и температуры и строгая пропорциональность сигнала детектора количеству вещества в широких пределах создали пламенно-ионизационному детектору репутацию лучшего универсального детектора.
Детектор по теплопроводности (катарометр)
Принцип действия детектора по теплопроводности (ДТП) основан на изменении температуры нагретых нитей (чувствительных элементов) в зависимости от теплопроводности газа, которая в свою очередь определяется его составом. ДТП измеряет различие в теплопроводности чистого газа-носителя и смеси газа-носителя с веществом, выходящим из хроматографической колонки. Поэтому наибольшая чувствительность может быть получена в том случае, когда теплопроводность анализируемого вещества сильнее отличается от теплопроводности газа-носителя. Большинство органических веществ имеют низкую теплопроводность, и для их анализа целесообразно использовать газы-носители с возможно более высокой теплопроводностью, например, гелий или водород.
7.
Вода — среда оптически более плотная, чем воздух. Если воду заменить какой-либо иной прозрачной средой, оптически более плотной, чем воздух, то преломлённый луч также будет приближаться к перпендикуляру. Поэтому можно сказать, что если свет идёт из среды оптически менее плотной в более плотную среду, то угол преломления всегда меньше угла падения (см. рис.):
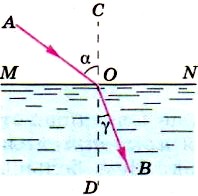 Рис. Схема преломления луча света при переходе из воздуха в воду
Рис. Схема преломления луча света при переходе из воздуха в воду
Луч света при переходе из воздуха в воду меняет своё направление, приближаясь к перпендикуляру CD.
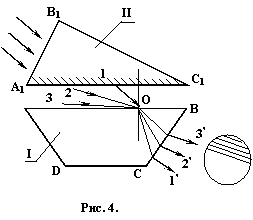
Основной узел рефрактометра АББЕ (модель ИРТ – 464) состоит из двух призм (рис. 4). Призма II с матовой гранью А1С1 является осветительной, а призма I с полированной гранью АВ – измерительной. Между призмами помещается исследуемая жидкость. Лучи света падают на грань А1В1, преломляются и попадают на матовую поверхность А1С1. Матовая поверхность рассеивает лучи, и в измеряемую жидкость входят лучи разных направлений, далее они проходят сквозь слой исследуемой жидкости и падают на поверхность АВ измерительной призмы. Если рассмотреть произвольную точку О на поверхности АВ, то лучи, входящие в измерительную призму в этой точке, имеют разные углы падения – от 0 до р/2. Так как жидкости, исследуемые с помощью рефрактометра, имеют показатель преломления меньше показателя преломления стекла измерительной призмы, то для каждого из этих лучей угол преломления будет меньше угла падения (r < i). При i= р/2 угол преломления достигает своего максимального значения rmax, которое, согласно закону Снеллиуса, равно
rmax = arcsin(![]() )<
)<![]() .
.
Так как ![]() , то при рассмотрении пучка лучей, выходящих из измерительной призмы в зрительную трубу, видно, что нижняя часть поля зрения трубы освещена, а верхняя остается темной. Граница светотени, наблюдаемая в окуляр, соответствует предельному углу преломления жидкости.
, то при рассмотрении пучка лучей, выходящих из измерительной призмы в зрительную трубу, видно, что нижняя часть поля зрения трубы освещена, а верхняя остается темной. Граница светотени, наблюдаемая в окуляр, соответствует предельному углу преломления жидкости.
8.
Кондуктометрический метод анализа основан на измерении электропроводности анализируемого раствора. Электропроводностью называют величину, обратную электрическому сопротивлению R. Единицей измерения электропроводности является Ом-1 или сименс (См). Растворы электролитов, являясь проводниками II рода, подчиняются закону Ома. По аналогии с сопротивлением проводников I рода, сопротивление раствора прямо пропорционально расстоянию между электродами l и обратно пропорционально площади их поверхности S
R = с(l / S),
где с - удельное сопротивление (Ом. см). При l = 1 см и S = 1 см2 имеем R= с, следовательно, удельное сопротивление равно сопротивлению 1 см3раствора, находящегося между двумя параллельными пластинами площадью 1 см2, отстоящими друг от друга на 1 см.
Величину, обратную удельному сопротивлению, называют удельной электропроводностью ч=1/с. Удельная электропроводность (См. см-1) численно равна току (в амперах) , проходящему через слой раствора с поперечным сечением, равным единице, под действием градиента потенциала 1 В на единицу длины.
Электропроводность разбавленных растворов электролитов зависит от числа ионов в растворе (т. е. от концентрации), числа элементарных зарядов, переносимых каждым ионом (т. е. от заряда иона), и от скорости движения одинаково заряженных ионов к катоду или аноду под действием электрического поля. С учетом всех этих факторов электропроводящие свойства ионов характеризуют эквивалентной ионной электрической проводимостью (подвижностью).
Эквивалентной электрической проводимостью называют проводимость раствора, содержащего 1 моль эквивалента вещества и находящегося между двумя параллельными электродами, расстояние между которыми 1 см. Ее единицей измерения является См. см2 . моль-1.
Удельная и эквивалентная проводимость связаны соотношением:
л= 1000 ч / с,
где с – молярная концентрация эквивалента, моль-экв/л.
Методы прямой кондуктометрии основываются на том, что в области разбавленных и умеренно концентрированных растворов электрическая проводимость растет с увеличением концентрации электролита.
9.
АТОМНО-АБСОРБЦИОННЫЙ АНАЛИЗ (атомно-абсорбц. спектрометрия), метод количеств. элементного анализа по атомным спектрам поглощения (абсорбции). Через слой атомных паров пробы, получаемых с помощью атомизатора (см. ниже), пропускают излучение в диапазоне 190-850 нм. В результате поглощения квантов света атомы переходят в возбужденные энергетич. состояния. Этим переходам в атомных спектрах соответствуют т. наз. резонансные линии, характерные для данного элемента. Согласно закону Бугера-Ламберта-Бера, мерой концентрации элемента служит оптич. плотность A = lg(I0/I), где I0 и I-интенсивности излучения от источника соответственно до и после прохождения через поглощающий слой
В зависимости от способа получения поглощающего слоя атомов выделяют 4 основных типов техники атомизации:
- пламенная атомизация – испарение и атомизация происходят в пламени (пропан/воздух, ацетилен/воздух, ацетилен/закись азота). Определяемые концентрации элементов в растворах 0,01 – 100 мг/л; электротермическая атомизация (ЭТА) – испарение и атомизация пробы происходит в графитовой трубке (графитовой печи), нагреваемой электрическим током до температур 1500 – 3000 °С (в зависимости от свойств определяемого элемента). Определяемые концентрации элементов в растворах 0,01 – 100 мкг/л; гидридная техника – в кварцевой ячейке или графитовой печи, нагреваемой электрическим током, происходит разложение газообразных гидридов, образованных в специальном реакторе: MeHxT --> Me + x/2 H2. Данная техника может использоваться для элементов, образующих термически неустойчивые газообразные гидриды (As, Sb, Se, Sn, Te, Pb). Определяемые концентрации элементов в растворах 0,01 – 100 мкг/л; метод «холодного пара» - основан на свойстве ртути существовать при нормальных условиях в газовой фазе в виде свободных атомов. Определяемые концентрации ртути в растворах 0,01 – 100 мкг/л.
Существует несколько видов источников света. Наиболее часто применяют лампы с полым катодом, безэлектродные лампы и настраивающиеся лазеры.
- Лампа с полым катодом состоит из полого катода цилиндрической формы, рядом с которым находится вольфрамовая проволока — анод. Сама лампа представляет собой цилиндрический стеклянный баллон, который наполнен инертным газом. Катод лампы изготовлен из определяемого в ходе анализа элемента или его сплава. Свет необходимой длины волны, поглощаемый в атомизаторе атомами определяемого элемента, в результате излучается. Наибольшая длина волны определяется линией Cs — 852 нм, наименьшая — линией As — 193,7 нм; более короткие волны в атомно-абсорбционном анализе не используют из-за сильного поглощения их кислородом воздуха. Внутри безэлектродной лампы с помощью катушки, по которой проходит ток высокой частоты, создается сильное электромагнитное поле. В это поле помещается маленькая кварцевая ампула, содержащая летучее соединение определяемого вещества. Принцип действия аналогичен принципу лампы с полым катодом. Основной недостаток такого вида источника света — необходимость в дополнительном устройстве для питания — высокочастотном генераторе Настраивающиеся лазеры в качестве источников излучения позволяет обойтись без большого набора ламп, так как один такой лазер можно использовать для всех элементов, однако широкому его использованию препятствует дороговизна
10.
Фотометрическое определение сахара
Фотоколориметрия может быть использована для количественного определения всех тех веществ, которые дают окрашенные растворы, или могут дать окрашенное растворимое соединение с помощью химической реакции.
Поскольку водный раствор, содержащий сахар, не окрашен, то прямо его фотометрировать нельзя. Но если предварительно добавить в раствор, содержащий сахар, какой – либо реагент, с которым сахар образует окрашенное соединение, то фотометрирование возможно. Известные методы:
Количественное определение сахаров по антроновому методу (с антроновым реактивом)
Количественное определение сахаров по реакции с пикриновой кислотой.
При взаимодействии редуцирующих сахаров с пикриновой кислотой они окисляются до соответствующих кислот, а пикриновая кислота восстанавливается в пикраминовую, обладающую красной или буровато-красной окраской:
Содержание сахара в испытуемом растворе рассчитывают по калибровочной кривой, составленной по стандартным (рабочим) растворам глюкозы (сахарозы). На оси абсцисс откладывают величины, характеризующие содержание
Рефрактометрический метод
Все природные моносахариды обладают оптической активностью. Их вращательная способность представляет собой не только важную константу, характеризующую эти соединения, но может быть также использована для количественного определения известных сахаров в растворе.
Чаще всего в контроле производства различных углеводов применяют поляриметрический метод, который основан на измерении угла вращения плоскости поляризации растворов сахаров. На результаты при определении сахаров этим методом влияют другие оптически активные вещества, которые затрудняют анализ смесей сахаров.
Данный метод годится только для определения высоких концентрация сахара. Если посмотреть справочные данные (справочник химика) по показателю преломления водных растворов сахаров, то увидим, какое незначительно изменение показателя преломления происходит в случае, когда концентрация раствора увеличивается на 1%:
1% nD20 1.33443
2% nD20 1.33588
Т. е. в нашем случае, когда концентрация сахара равна 0.01% при построении градуировочного графика на столь малых концентрациях показатель преломления практически не будет меняться, и стало быть, не будет функциональной зависимости показателя преломления от концентрации. Т. е. метод рефрактометрии не годится для определения столь малых концентрация сахара.
11.
Радиоактивационные методы анализа - метод качественного и количественного элементного анализа вещества, основанный на активации ядер атомов и исследовании образовавшихся радиоактивных изотопов(радионуклидов). Вещество облучают ядерными частицами (тепловыми или быстрыми нейтронами, протонами, дейтронами, б-частицами и т. д.) или г-квантами. Затем определяют вид, т. е. порядковый номер и массовое число, образовавшихся радионуклидов по их периодам полураспада Т1/2 и энергиям излучения Е, которые табулированы. Поскольку ядерные реакции, приводящие к образованию тех или иных радионуклидов, обычно известны, можно установить, какие атомы были исходными.
Количественный активационный анализ основан на том, что активность образовавшегося радионуклида пропорциональна числу ядер исходного изотопа, участвовавшего в ядерной реакции.
Абс. метод характеризуется высокой погрешностью (относит. стандартное отклонение 0,4-0,6), что связано с неконтролируемыми колебаниями величины Ф, сложностью определения E, погрешностями табличных значений а и т. д. Поэтому обычно анализ выполняют относит. методом, основанным на сравнении активностей анализируемого образца и образцов сравнения с точно известным содержанием определяемых элементов. Облучение и измерение активности образцов проводят в одинаковых условиях.
Существуют два основных варианта активационного анализа - инструментальный и радиохимический. Первый применяют при анализе веществ, которые либо слабо активируются, либо образуют короткоживущие радионуклиды. Анализируемый образец и образцы сравнения одновременно получают и затем обычно несколько раз измеряют (с помощью полупроводникового спектрометра высокого разрешения) и сопоставляют ихг - спектры. При первом измерении идентифицируют и определяют содержание элементов, образующих короткоживущие радионуклиды, при втором-элементы, образующие радионуклиды с большим Т1/2, и т. д. Кроме того, последоват. измерение г - спектров позволяет идентифицировать радионуклиды не только по энергиям испускаемых г - квантов, но и по T1/2.
Основные достоинства инструментального варианта: быстрота проведения, сравнительно небольшая трудоемкость, высокая информативность, возможность проводить анализ без разрушения образца и использовать радионуклиды с небольшими Т1/2 (от нескольких минут до нескольких секунд). Широкое использование электронно-вычислит. техники для оптимизации условий анализа и обработки спектрометрической информации повысило точность и надежность метода и позволило создать полностью автоматизированные системы активационного анализа. Основной недостаток инструментального варианта: невозможность анализировать сильно активируемые вещества, образующие долгоживущие радионуклиды.
В радиохимическом варианте облученный образец растворяют, а затем отделяют от основы образовавшиеся радионуклиды определяемых элементов, обычно вместе с их изотопными носителями (неактивными изотопами), которые специально добавляют в р-р. Методы разделения-экстракция, хроматография, дистилляционные методы и др.; они позволяют получать препараты определяемых элементов радиохим. степени чистоты, активность которых можно измерять на полупроводниковом спектрометре. При доминирующем содержании одного или неск. элементов прямой гамма-спектральный анализ затруднен и необходимо эти радионуклиды разделять на группы, удобные для измерения г - спектров. Для достижения особенно низких пределов обнаружения выделяют индивидуальные элементы.
Наиб. распространен нейтронно-активационный анализ, в к-ром исследуемое в-во облучают потоком тепловых нейтронов с энергией 0,025 эВ, т. к. сечения ядерных реакций в этом случае для большинства элементов на неск. порядков выше сечений др. ядерных р-ций. Поток нейтронов из ядерных реакторов достигает 1013-1015 частиц/см2*с. Метод позволяет определять большинство элементов периодич. системы начиная с Na с пределами обнаружения 10-4 - 10-12%, в т. ч. 53 элемента - с пределами обнаружения менее 10-6%
Достоинства активационног анализа: высокая чувствительность, возможность в ряде случаев проводить определение без разрушения образца, высокая избирательность, возможность одновременного определения ряда примесей в одной навеске образца, отсутствие поправки контрольного опыта (т. к. все хим. операции, в т. ч. травление образцов для удаления поверхностных загрязнений, проводят после облучения). Кроме того, при работе с короткоживущими радионуклидами анализ м. б. выполнен быстро-в течение неск. минут. Недостатки метода: относительно малая доступность источников ядерных частиц или г--квантов, возможность деструкции и даже разрушения образцов при облучении мощными потоками излучений, относит. сложность выполнения анализа, радиац. опасность.
Основные области применения активационного анализа: анализ особо чистых веществ, геол. объектов и объектов окружающей среды; экспрессный анализ металлов и сплавов в пром-сти; определение содержания микроэлементов в крови, плазме, тканях животных и растений; судебно-мед. экспертиза.
