


Партнерка на США и Канаду по недвижимости, выплаты в крипто
- 30% recurring commission
- Выплаты в USDT
- Вывод каждую неделю
- Комиссия до 5 лет за каждого referral
, ,
Оптимизация аналитических методов обнаружения, идентификации и количественного определения охратоксина А в пищевых продуктах.
ГУ НИИ питания РАМН, Москва
Охратоксин А - вторичный метаболит широко распространенных микроскопических плесневых грибов родов Penicillium (P. verrucosum) и Aspergillius (A. ochraceus) стоит в ряду приоритетных микотоксинов – контаминантов пищевых продуктов, и представляет реальную опасность для здоровья человека. Охратоксин А обладает выраженным нефротоксическим, канцерогененным, а также тератогененным, иммунотоксическим, нейротоксическим, генотоксическим и цитотоксическим действием [2, 19, 20]. Международным агентством по исследованию рака (IARC) охратоксин А отнесен к веществам, возможно канцерогенным для человека (группа 2В) [9]. При введении внутрь LD50 варьирует для разных видов животных от 20-30 мг/кг м. т. (для крыс) до 1 мг/кг м. т. (для свиней) [19]. Охратоксин А рассматривается в качестве одного из этиологических факторов Балканской эндемической нефропатии - тяжелого почечного заболевания, регистрируемого в некоторых восточноевропейских странах (в частности Болгарии, Румынии, Сербии, Хорватии, Боснии и Герцеговине, Словении, Македонии) [12, 19]. Охратоксин А наиболее часто обнаруживается в зерновых продуктах, кофе, специях. В последнее время появляются данные о значительном загрязнении охратоксином А сухофруктов, вина и фруктовых соков. Так, в результате исследований, проведенных в странах ЕС, было установлено, что около 70% партий зерновых продуктов содержали охратоксин А в диапазоне от 0,00001 до 0,041 мг/кг, около 60% исследованных партий вина были загрязнены охратоксином А в количестве от 0,000003 до 0,016 мг/л [18]. Особого внимания заслуживают данные о частом обнаружении охратоксина А в крови, а также материнском молоке населения многих европейских стран, что свидетельствует о постоянном поступлении этого микотоксина в организм человека [18, 19, 22]. Основной вклад в величину поступления охратоксина А с продуктами питания вносят зерновые (44%), вино (10%) и кофе (9%) [18]. Рекомендуемое Объединенным комитетом экспертов ФАО/ВОЗ по пищевым добавкам (JECFA) допустимое недельное потребление охратоксина А составляет 100 нг/кг/м. т. [19]. Содержание охратоксина А в странах ЕС регламентируется на уровне 0,005 мг/кг в продовольственном сырье и 0,003 мг/кг в продуктах питания [8]. Достоверные данные о загрязнении пищевых продуктов охратоксином А в РФ отсутствуют.
Разработанный ранее для нужд санитарно-эпидемиологической службы СССР метод определения охратоксина А в пищевых продуктах [1], использующий жидко-жидкостное распределение для очистки экстракта, имеет ряд недостатков: относительно низкая чувствительность метода (предел обнаружения - 0,001-0,002 мг/кг), значительная продолжительность анализа, а также необходимость использования большого количества хлорсодержащих органических растворителей.
К настоящему времени разработаны новые более эффективные методы определения охратоксина А в пищевых продуктах, основанные на применении твердофазной экстракции (ТФЭ) [18, 19]. Для ТФЭ характерна хорошая очистка экстракта, высокая степень извлечения и малый расход растворителей. При ТФЭ используют нормально-фазовую адсорбционную хроматографию на силикагеле [14, 19], обращено - фазовую распределительную хроматографию на силикагеле, химически модифицированном октадецилсиланом [4, 11], иммуноаффинную хроматографию [3, 23], а также колоночную хроматографию на других сорбентах (диатомитовая земля (целит), полиамид, полимеры, полученные методом импринтинга и др.) [5, 7, 10, 15].
Слабокислотные свойства (рКА = 4,4) охратоксина А (рис.1) определяют необходимость его экстракции либо в недиссоциированной форме смесями органических растворителей с кислотными водно-солевыми растворами [1, 18, 19], либо в виде соли - водными слабощелочными растворами, например, раствором гидрокарбоната натрия [6, 21].
Обращенно-фазовая ВЭЖХ (ОФ ВЭЖХ) с флуориметрическим детектированием является наиболее широко распространенным методом определения охратоксина А в пищевых продуктах [1, 4, 13, 16, 18, 23, 24].
Целью исследования явилась разработка чувствительного метода обнаружения, идентификации и количественного определения охратоксина А в пищевых продуктах путем оптимизации аналитических подходов, основанных на использовании ТФЭ.
Экспериментальная часть
Аппаратура и реагенты. Хроматографическая система состояла из насоса высокого давления Jasco 880-PU, инжектора Rheodyne-7125 с объёмом петли-дозатора 20 мкл, флуориметрического детектора FL Detector model LC305 (Linear Instruments) (лвозб.=250 нм, лэмис.= 458 нм) и системы для сбора и обработки данных «Мультихром» (Амперсенд). Хроматографическая колонка (250 * 4,6 мм) с неподвижной фазой Kromasil C18 (MetaChem Technologies Inc.), с размером частиц 5 мкм.
Экстракцию проводили с использованием аппарата для встряхивания shaker s-3.08L (ELMI), для центрифугирования проб использовали центрифугу ЦЛС 31М. Твёрдофазную экстракцию проводили с использованием вакуумного манифолда Macherey-Nagel, патронов ДИАПАК Силикагель (БиоХимМак СТ), иммуноаффинных колонок (ИАК) OCHRAPREP (R-BIOFARM RHONE LTD). рН растворов измеряли рН-метром МР 230 (Mettler Toledo). Концентрирование проб проводилось на роторном испарителя Laborota 4000 (Heidolph). Для растворения стандартов и упаренных экстрактов использовалась ультразвуковая ванна УЗВ-12л (ПКФ САПФИР). Для подбора оптимальных волн возбуждения и эмиссии использован флуоресцентный спектрофотометр Cary Eclipse (Varian). В качестве стандарта использовался стандартный образец охратоксина А в смеси бензол-уксусная кислота (99:1) с концентрацией С = 9,2 нг/мкл.
Твердофазная экстракция:
- Очистка с помощью колоночной хроматографии (КХ) на диатомитовой земле. Экстракцию охратоксина А из 25,0 г. измельченной пробы проводили 125 мл хлороформа после добавления 20 мл 2% уксусной кислоты. Перемешивали на аппарате для встряхивания в течение 30 минут. 50 мл пропущенного через бумажный фильтр хлороформного экстракта наносили на колонку с диатомитовой землей, импрегнированной гидрокарбонатом натрия. Колонку промывали последовательно 70 мл гексана и 30 мл хлороформа. Охратоксин А элюировали с колонки 150 мл смеси бензол – ацетон – уксусная кислота (86:12:2). Элюат упаривали досуха, растворяли в 3 мл подвижной фазы (ацетонитрил – водная Н3РО4 (рН=2.6) (62:38) с использованием ультразвуковой ванны.
- Очистка с помощью КХ на силикагеле. Экстракцию охратоксина А из 20,0 г. измельченной пробы проводили 100 мл толуола после последовательного добавления 30 мл 2М раствора соляной кислоты и 50 мл 0,4М раствора хлорида магния. Перемешивали 60 минут, центрифугировали со скоростью 3500 об./мин 5 минут. Верхний толуоловый слой фильтровали через бумажный фильтр, отбирая 50 мл фильтрата. После кондиционирования 10 мл толуола на патрон с силикагелем наносили 50 мл фильтрата, промывали дважды 10 мл н-гексана и 10 мл смеси толуол – ацетон (85:15), затем 5 мл толуола. Охратоксин А элюировали 40 мл смеси толуол – ацетон – уксусная кислота (89:10:1). Элюат упаривали досуха, растворяли в 1 мл подвижной фазы (ацетонитрил – водная Н3РО4 (рН=2.6) (62:38) с использованием ультразвуковой ванны.
-Очистка с помощью иммуноаффинной КХ [17]. Экстракцию охратоксина А из 50,0г. измельченной пробы проводили 200 мл смеси ацетонитрил – вода (60:40). Перемешивали в течение 30 минут. 4 мл пропущенного через бумажный фильтр экстракта смешивали с 44 мл фосфатного буфера и наносили на иммуноаффинную колонку (ИАК), которую затем промывали 20 мл фосфатного буфера. Остатки фосфатного буфера удаляли продуванием ИАК воздухом. Охратоксин А элюировали 3 мл смеси метанол – уксусная кислота (98:2). Элюат упаривали досуха, растворяли в 1 мл подвижной фазы (ацетонитрил – водная Н3РО4 (рН=2.6) (62:38) с использованием ультразвуковой ванны.
Условия ВЭЖХ. Идентификацию и количественное определение охратоксина А проводили методом ОФ ВЭЖХ в режиме изократического элюирования с флуоресцентным детектированием (лвозб.=250 нм, лэмис.=458 нм). В качестве подвижной фазы использовали смесь ацетонитрил – водная Н3РО4 (рН=2.6) (62:38). Скорость элюирования составляла 1 мл/мин. В систему для ВЭЖХ вводилось 20 мкл исследуемого раствора.
Результаты и их обсуждение.
При очистке экстракта путем КХ с диатомитовой землей, импрегнированной гидрокарбонатом натрия, в качестве базового был использован метод АОАС 975.38 [5], предусматривающий применение ТСХ для идентификации и количественного определения охратоксина А. Нами в целях повышения чувствительности и специфичности метода была использована ОФ ВЭЖХ с флуоресцентным детектированием. В результате применения базового метода степень извлечения охратоксина А из матрикса, искусственно контаминированного охратоксином А на уровне 0,01 мг/кг, в наших условиях не превысила 40%. В целях повышения величины извлечения нами был внесен целый ряд изменений в условия экстракции и очистки: в состав экстрагирующей смеси была добавлена уксусная кислота; объем хлороформа, используемого для промывания колонки, уменьшен до 30 мл; в состав элюирующей смеси внесен ацетон; объем элюирующей смеси увеличен до 150 мл. В результате оптимизации метода величина извлечения охратоксина А из пшеницы возросла до 60% (табл.1).
Степень извлечения удалось существенно повысить, применив метод твердофазной экстракции на патронах с немодифицированным силикагелем (схема, приближенная к методу ICC № 000 [14]). Корректировка базового метода [14] (увеличено содержание толуола в смеси толуол – ацетон, используемой для промывания колонки, до 85%, в состав элюирующей смеси внесен ацетон, объем элюирующей смеси доведен до 40 мл) позволила увеличить степень извлечения до 80%.
При использовании иммуноаффинной КХ наблюдалась максимальная степень извлечения охратоксина А из пищевого матрикса (до 100%), При этом предел обнаружения (0,0005 мг/кг) был выше, чем при очистке КХ на силикагеле (0,00005 мг/кг).
Для обнаружения, идентификации и количественного определения охратоксина А с помощью ВЭЖХ был подобран оптимальный состав подвижной фазы (ПФ) и уточнены длины волн возбуждения и эмиссии флуоресценции, обеспечившие максимальный сигнал и селективность при детектировании охратоксина А (табл.2). Подобранная длина волн возбуждения (250 нм вместо 333 нм) и эмиссии флуоресценции для оптимизированной подвижной фазы позволила повысить отношение сигнал/шум с соответствующим снижением предела обнаружения метода. Изменение состава ПФ позволило улучшить разделение пиков охратоксина А и компонентов матрикса пищевого продукта при одновременном снижении времени удерживания пика охратоксина А (рис.2).
Возможность использования разработанного нами модифицированного метода, основанного на применении патронов с силикагелем, в рутинном анализе охратоксина А была подтверждена выборочным исследованием частоты и уровня загрязнения этим микотоксином основных видов продовольственного сырья. Для этого было изучено продовольственное зерно урожая 2004 года из различных регионов РФ (табл.3). Девять из 46 исследованных образцов зерна содержали охратоксин А в диапазоне от 0,00005 до 0,005 мг/кг, что не превышало регламенты, принятые в странах ЕС.
Таким образом, путем оптимизации существующих аналитических подходов предложены два варианта метода обнаружения, идентификации и количественного определения охратоксина А в пищевых продуктах: с использованием КХ на силикагеле или иммуноаффинной КХ для очистки экстрактов и оптимизированных условий ВЭЖХ. Метод, основанный на применении КХ на силикагеле, отличается низким пределом обнаружения и невысокой стоимостью расходных материалов. Очистка с помощью иммуноаффинной КХ обеспечивает высокую степень извлечения и чистоту экстракта, а также характеризуется более высоким пределом обнаружения (0,0005 мг/кг вместо 0,00005 мг/кг для варианта с очисткой КХ на силикагеле). Полученные в результате проведенного выборочного исследования содержания охратоксина А в продовольственном сырье данные свидетельствуют о необходимости дальнейшего изучения загрязнения охратоксином А продуктов питания в целях оценки риска контаминации этим микотоксином пищевых продуктов для здоровья населения РФ.
Автор для переписки:
ГУ НИИ питания РАМН
109240, Москва, Устьинский проезд, д.2/14
298-18-67
I. V. Aksenov, K. I. Eller, V. A. Tutelyan
Optimization of analytical methods for ochratoxin A analysis in food.
Ochratoxin A is a mycotoxin produced by widely distributed Aspergillus and Penicillium species. The mycotoxin is a common contaminant of cereals, coffee, wine, dried fruits and spices. Ochratoxin A has been shown to be nephrotoxc, immunosuppressive, embryotoxic, teratogenic and carcinogenic in many mammalian species. Codex Alimentarius and EC have established maximum permissible level of 5 мg/kg for ochratoxin A in raw cereal grains and of 3 мg/kg – for ready-to-eat products derived from cereals. Two simple and reliable modifications of methods have been developed for the analysis of ochratoxin A in food which are based on immunoaffinity or silica gel column clean-up and HPLC with fluorescence detection. The detection limits were 0,5 мg/kg and 0,05 мg/kg respectively. Methods have been used successfully to analyze ochratoxin A in 46 samples of raw cereals harvested in different regions of Russia. Nine samples were found to be contaminated with ochratoxin A on levels ranged from 0,05 to 5 мg/kg.
, ,
Оптимизация аналитических методов обнаружения, идентификации и количественного определения охратоксина А в пищевых продуктах.
Охратоксин А – микотоксин, продуцируемый широко распространенными плесневыми грибами родов Aspergillius и Penicillium. Он является частым контаминантом зерновых продуктов, кофе, вина, сухофруктов и специи. Доказано нефротоксическое, иммуносупрессивное, эмбриотоксическое, тератогенное и канцерогенное действие охратоксина А для многих видов млекопитающих. Установленный Кодексом Алиментариус и ЕС максимальный допустимый уровень содержания охратоксина А в зерне равен 5 мкг/кг, в готовых к употреблению зерновых продуктах – 3 мкг/кг. Предложены два оптимизированыx метода обнаружения, идентификации и количественного определения охратоксина А в пищевых продуктах: с использованием КХ на силикагеле или иммуноаффинной КХ для очистки экстрактов и ВЭЖХ с флуоресцентной детекцией. Предел обнаружения составил соответственно 0,05 и 0,5 мкг/кг. Разработанные методы использованы для анализа содержания охратоксина А в 46 образцах зерна, собранного в различных регионах России. Девять образцов были загрязнены охратоксином А в количестве от 0,05 до 5 мкг/кг.
Подписи к рисункам.
Рисунок 1. Химическая структура охратоксина А.
Рисунок 2. Хроматограммы образца пшеницы, контаминированного охратоксином А на уровне 0,01 мг/кг, при различных способах очистки:
А) КХ на диатомитовой земле Б) КХ на силикагеле В) иммуноаффинная КХ.
Рис. 1.
Рис. 2.
А
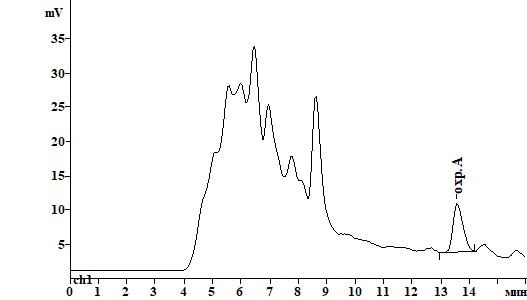
Б
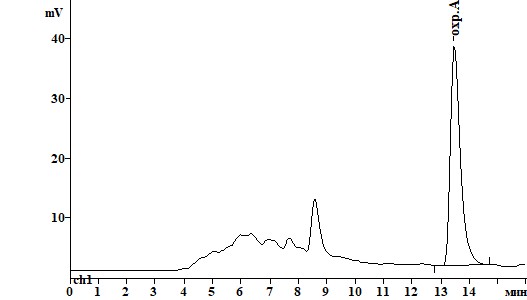
В
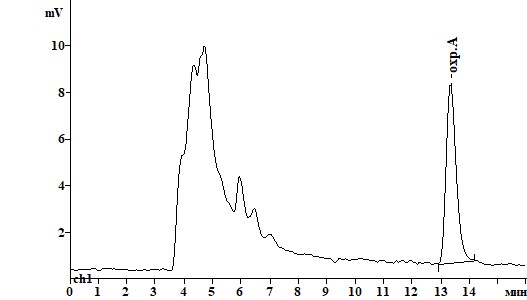
Таблица 1. Сравнительная характеристика различных способов очистки экстракта.
Способ ТФЭ | Экстракция | Степень извлечения, % | Предел обнаружения, мг/кг |
КХ на диатомитовой земле | 125 мл хлороформа 20 мл 2% укс. к-ты | 40-60 | 0,0005 |
КХ на силикагеле | 100 мл толуола 30 мл 2М соляной кислоты 50 мл 0,4М хлорида магния | 75-80 | 0,00005 |
Иммуноаффинная КХ | 200 мл ацетонитрил – вода (60:40) | 95-100 | 0,0005 |
Таблица 2. Сравнительная характеристика параметров ВЭЖХ (для 1 нг охратоксина А во вколе).
Метод | Состав ПФ | Длины волны флуориметрического детектированния, нм | Время удерживания охр. А, мин | Отношение сигнал/шум | |
лвозб | лэмис. | ||||
[14] | ацетонитрил – вода – уксусная кислота (99:99:2) | 333 | 460 | 27-29 | 230 |
ацетонитрил – вода – уксусная кислота (99:99:2) | 250 | 460 | 27-29 | 390 | |
[17] | ацетонитрил – вода – уксусная кислота (102:94:4) | 333 | 443 | 24-25 | 120 |
ацетонитрил – вода – уксусная кислота (102:94:4) | 250 | 443 | 24-25 | 220 | |
Оптимизи-рованная методика | ацетонитрил – водная Н3РО4 (рН=2.6) (124:76) | 250 | 458 | 13-15 | 430 |
ацетонитрил – водная Н3РО4 (рН=2.6) (124:76) | 333 | 458 | 13-15 | 290 |
Таблица 3. Выборочное исследование уровня загрязнения охратоксином А продовольственного зерна урожая 2004 г.
Количество партий | Количество партий, содержащих охратоксин А | Содержание охратоксина А в контаминированных пробах, мг/кг | |
пшеница | 32 | 2 | 0,00007 и 0,0003 |
рожь | 6 | 3 | 0,00019-0,00101 |
ячмень | 8 | 4 | 0,00005-0,00506 |
ВСЕГО | 46 | 9 | 0,00005-0,00506 |
Литература
Методические рекомендации по обнаружения, идентификации и определению содержания охратоксина А в пищевых продуктах. - М., 1985. , Кравченко (Медицинские и биологические аспекты) – М., 1985. AOAC, Determination of Ochratoxin A in Wine and Beer 2001.01 // J. AOAC Int. – 2001. - Vol. 84. - P. 1818. AOAC, Ochratoxin A in Corn and Barley 991.44 // J. AOAC Int. – 1996. - Vol. 79. – P. 1102-1105. AOAC, Ochratoxin A in Green Coffee 975.38 // J. AOAC Int. – 1975. - Vol. 58. - P. 258. Application note for analisis of ochratoxin A in cereal using sodium bicarbonate extracton in conjunction with Ochraprep. – Glasgow, 2001. Baggiani C., Giraudi G., Vanni A. // Bioseparation. - 2002. - Vol.10. – P. 389–mission Regulation (EC) № 000/2002 // Official Journal of the European Communities. - 2002. - L 75. - P. 18-20. IARC Monographs on the Evaluation of Carcinogenic Risks to Humans. Vol. 56. – Lion, 1993. Jodlbauer J., Maier N. M., Lindner W. // Journal of Chromatography A. - 2002. - Vol. 945. – P. 45–63. Jornet D., Busto O., Guasch J // Journal of Chromatography A. - 2000. - Vol. 882. – P. 29–35. Krogh P. // Endemic nephropathy, Proceedings of the second International Symposium on Endemic nephropathy 9-12 November 1972. - Sofia, 1972. – P. 266-277. Kuhn I., Valenta H., Rohr K. // Journal of Chromatography B. - 1995. - Vol. 668. – P. 333–337. Majerus P., Weber R., Wolff J. // Bundesgesundheitsblatt. – 1994. – B. 37, N. 11. – S. 454 – 458. Monaci L., Palmisano F.// Anal. Bioanal. Chem. - 2004. - Vol. 378. - P. 96-103. Monaci L., Tantillo G., Palmisano F. // Anal. Bioanal. Chem. - 2004. - Vol. 378. – P. 1777–1782. Quantitative detection of Ochratoxin A.- Glasgow, 2003. Report of experts participating in Task 3.2.7 “Assessment of dietary intake of Ochratoxin A by the population of EU Member States”. – Rome, 2002. Safety evaluation of certain mycotoxins in food. // WHO Food Additives Series, No.47; FAO Food and Nutrition Paper 74. – Geneva, 2001. Schwartz G. G. // Cancer Causes Control. - 2002. - Vol.13. - P. 91-100. Scott P. M. // Adv. Exp. Med. Biol. - 2002. - Vol. 504. – P. 117-134. Skaug MA, Helland I, Solvoll K, Saugstad O. D. // Food Addit. Contam. - 2001. - Vol. 18. – P. 321-327. Visconti A., Pascale M., Centonze G. // Journal of Chromatography A. - 1999. - Vol. 864. – P. 89–101. Zimmerli B., Dick R. // Journal of Chromatography B. – 1995. - Vol. 666. – P. 85 – 99.