


Партнерка на США и Канаду по недвижимости, выплаты в крипто
- 30% recurring commission
- Выплаты в USDT
- Вывод каждую неделю
- Комиссия до 5 лет за каждого referral

Московский Государственный университет
им.
Факультет фундаментальной физико-химической инженерии
Институт проблем химической физики РАН
Лаборатория ионики твёрдого тела
Определение удельной площади поверхности платины, нанесённой на оксидный носитель.
Курсовая работа по аналитической химии
студента 2 курса
Научный руководитель
к. х.н,
Москва, 2016
Оглавление
1.Введение 2
2. Литературный обзор. 3
2.1. Методы определения удельной площади поверхности. 3
2.1.1. Метод Брунауэра-Эммета-Теллера (БЭТ) 3
2.1.2. Десорбция водорода. 4
2.1.3. Десорбция металлов. 5
2.1.4. Десорбция монооксида углерода. 7
2.2. Особенности определения площади поверхности платины на оксидных носителях. 8
2.2.1. Спилловер. 8
2.2.2. Растворение оксидов 9
2.3. Вывод. 10
3. Методика исследований. 11
3.1. Подготовка рабочего электрода. 11
3.2. Электрохимическая ячейка. 11
3.3. Режим измерений. 12
4. Результаты и их обсуждение. 13
5. Заключение. 17
6.Список литературы. 18
1.Введение
По целому ряду причин платина является основой многих электрохимических элементов, однако свойства такой системы сильно зависят от среды, в которую она помещена. Решением этой проблемы может стать использование платины, нанесенной на подложку, которая может изменить свойства системы. Правильно подобранный носитель предотвращает агломерацию активного компонента, инертен в условиях катализа, обладает сильно развитой поверхностью и положительным образом влияет на химические свойства системы. Для электрокаталитичеких реакций большое значение имеет удельная проводимость и адсорбционная способность вещества-подложки.
Особый интерес в качестве носителя платины вызывает TiO2 допированный RuO2. Использование данной подложки способствует повышению толерантности к монооксиду углерода[1] (главному каталитическому яду платины), увеличивает проводимость, позволяет создать большую поверхность раздела фаз (что особенно важно в гетерогенном катализе), а также, поскольку сам по себе диоксид титана обладает значительной каталитической активностью, для данной системы могут наблюдаться различные суммарные эффекты[2].
Для корректного описания и сравнения образцов требуется знать удельную площадь поверхности Pt, однако классические методы для оксидных носителей активного вещества имеют ряд ограничений.
Целью настоящей работы является выявление оптимальных методик и условий для определения удельной площади поверхности платины на твёрдом растворе RuO2 в TiO2.
2. Литературный обзор.
2.1. Методы определения удельной площади поверхности.
2.1.1. Метод Брунауэра-Эммета-Теллера (БЭТ)
Суть метода заключается в следующем: вначале поверхность рассматриваемых материалов освобождается от адсорбированных на них веществ путем нагрева (проводится термотренировка образца). Затем вблизи температуры кипения адсорбата поверхность покрывают инертным газом: обычно азот, аргон или криптон.
Зная количество газа, адсорбированного на единице массы контролируемых материалов, а также кинетические диаметры молекул газа, можно рассчитать удельную поверхность по уравнению БЭТ. Погрешность полученных этим методом данных по площади удельной поверхности составляет около 10% от реальных величин.
Описанный выше метод имеет множество положительных сторон, но он не подходит для определения площади металлов, помещенных на носитель. Еще одним фактором, ограничивающим применение данного метода, является агломерация наночастиц платины при термотренировке образца.
2.1.2. Десорбция водорода.
Для электродов из платины (а так же Rh, Ir, Ni) электрохимическая адсорбция водорода из раствора происходит в виде монослоя в области потенциалов недонапряжения[3]. Определение количества адсорбированного водорода проводят методом циклической вольтамперометрии. Площадь под вольтамперометрическим пиком десорбции (исправленная на заряжение двойного электрического слоя) пропорциональна заряду, соответствующему монослойному заполнению, а, следовательно, и удельной поверхности платины. Истинную электроактивную поверхность электрода рассчитывают по формуле S = QH/QS, где QH – заряд, соответствующий монослойной заполнении поверхности; QS – заряд, соответствующий осаждению водорода на единицу площади поверхности. Для поликристаллической платины QS = 210 мкКл/см2 [3].
Данный метод широко используется для определения истинной площади поверхности мелкодисперсной Pt на различных носителях, является наиболее экспрессным из кулонометрических, но имеет ряд существенных недостатков: 1) высота и положение вольтамперометрических пиков зависит от природы электролита; 2) имеется сложность разделения фарадеевского и емкостного зарядов по вольтамперограмме (рис.1); 3) трудность определения конечной точки адсорбции монослоя. Ошибка определения может составить до 30% от истинного значения.
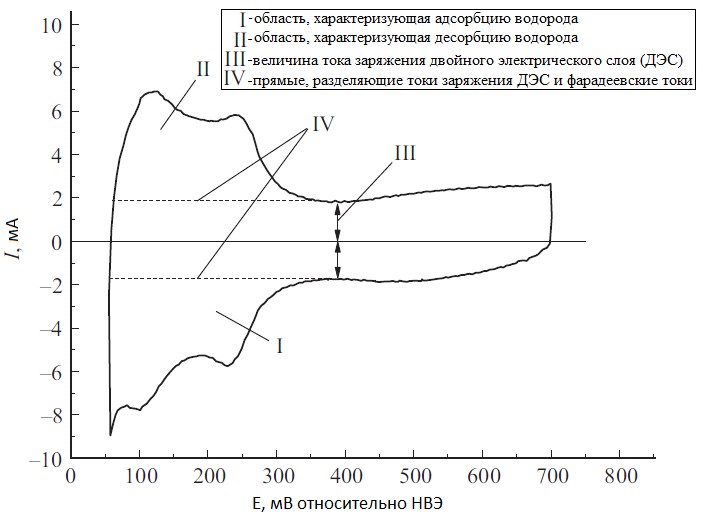
Рисунок №1. Характерная циклическая вольтамперограмма наноструктурированной Pt на углеродной саже в системе H2/N2.[4]
2.1.3. Десорбция металлов.
Некоторые металлы при потенциалах, превышающих потенциал равновесного осаждения металла, могут осаждаться на поверхности других металлов в отношении 1:1. В случае платинированных образцов диоксида титана допированного диоксидом рутения, медь является наиболее подходящим адатомом из-за сходства атомных радиусов металлов (Cu 128 пм; Pt 138,5 пм). Адсорбцию меди осуществляют в потенциостатическом режиме для достижения условий, наиболее приближенных к монослойному заполнению поверхности Pt. Заряд, соответствующий монослойному осаждению меди, рассчитывают, интегрируя пик десорбции меди (за вычетом базовой линии) на циклической вольтамперограмме: QM = ![]()
![]() где I-сила тока; Ich - сила тока заряжения двойного электрического слоя. Площадь поверхности рассчитывают по формуле S = QM/QS, где QS – заряд, соответствующий осаждению адатомов на единицу площади поверхности. Для осаждения меди на поликристаллическую платину QS = 420 мкКл/см2, (рассчитывается по кристаллографическим параметрам)[3]. Это число в 2 раза превышает аналогичную величину для водородного метода, так как процесс осаждения адатомов меди на поверхность платины сопровождается двухэлектронным переходом: Pt + Cu2+ + 2e = Pt-Cu.
где I-сила тока; Ich - сила тока заряжения двойного электрического слоя. Площадь поверхности рассчитывают по формуле S = QM/QS, где QS – заряд, соответствующий осаждению адатомов на единицу площади поверхности. Для осаждения меди на поликристаллическую платину QS = 420 мкКл/см2, (рассчитывается по кристаллографическим параметрам)[3]. Это число в 2 раза превышает аналогичную величину для водородного метода, так как процесс осаждения адатомов меди на поверхность платины сопровождается двухэлектронным переходом: Pt + Cu2+ + 2e = Pt-Cu.
На циклической вольтамперограмме меди (рис.2) можно выделить характерные пики: 1 соответствует десорбции фазовой меди, 2 – десорбции адатомов меди, 3 – адсорбции адатомов меди и 4 – адсорбции объёмной меди.
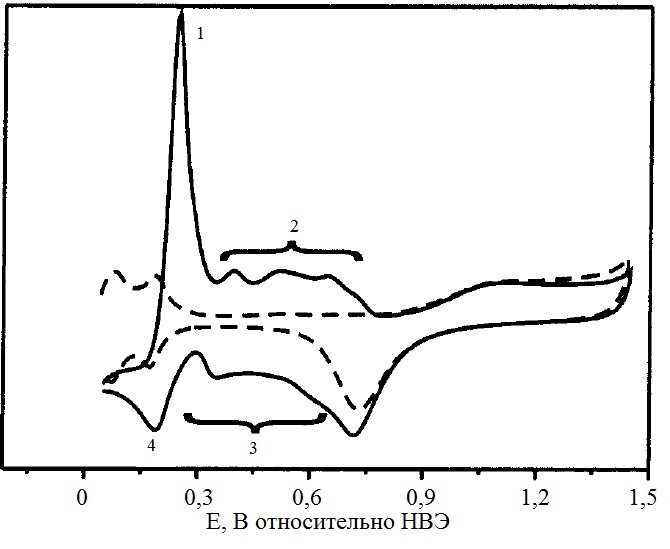
Рисунок №2. Характерная циклическая вольтамперограмма платинового электрода в растворе 0,1М H2SO4 и 0,5мМ CuSO4. Скорость развертки 0,01 В/с[5].
2.1.4. Десорбция монооксида углерода.
Монооксид углерода адсорбируется в качестве монослоя на электроды, состоящие из платины или некоторых сплавов на её основе [5]. Благодаря этому можно проводить измерения по описанной ранее методике десорбции водорода, используя в качестве рабочего газа CO.
Поскольку монооксид углерода – двухатомный газ, он может адсорбироваться на платину двумя способами: линейным и мостиковым, занимая соответственно один или два атома Pt. Это может отражаться на вольтамперограмме образованием перед основным пиком, который соответствует мостиковому расположению адсорбированного CO, предпика соответствующего линейному способу адсорбции молекул. Константа пересчета для монооксида углерода, десорбированного из мостиковой формы QS = 420 мкКл/см2, а из линейной QS = 210 мкКл/см2.
Использование монооксида углерода позволяет облегчить разделение фарадеевского и емкостного зарядов при обработке циклических вольтамепограмм (рис.3).
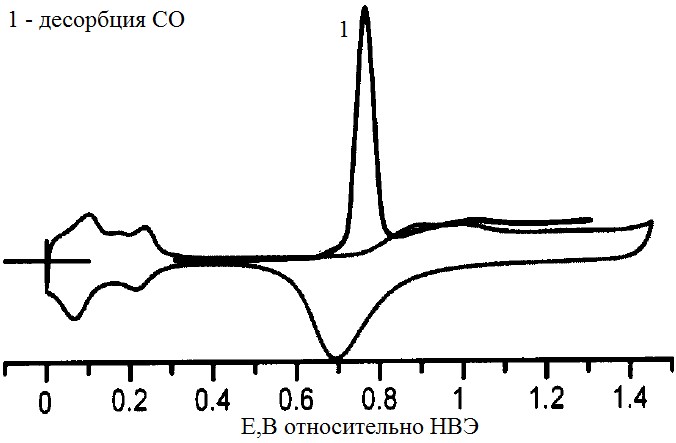
Рисунок №3. Характерная циклическая вольтамперограмма платинового электрода в растворе, насыщенном монооксидом углерода[5].
2.2. Особенности определения площади поверхности платины на оксидных носителях.
2.2.1. Спилловер.
Спилловером называют транспорт активных частиц, сорбированных или образованных на одной фазе, которая в данных условиях не сорбирует или не образует эти частицы (рис.4) [6]. Фаза, генерирующая активные частицы, носит название инициатора, а фаза, предоставляющая места для адсорбции активных частиц - акцептора. Это явление благоприятно сказываться на каталитических способностях многофазных систем, что объясняется протеканием одной из стадий сложного процесса на одной фазе, другой стадии - на другой, а между фазами осуществляется спилловер (иногда с участием
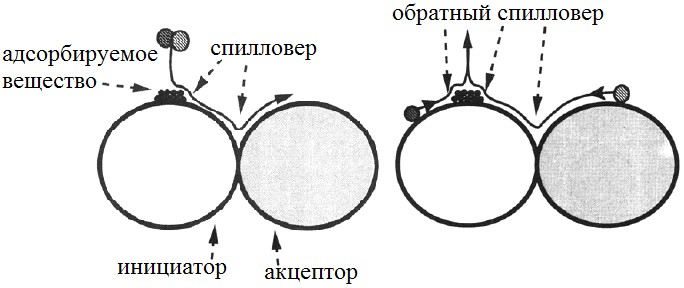
третьей фазы) [7].
Рисунок №4. Схема прямого и обратного спилловера[8].
Для платинированного диоксида титана, допированного диоксидом рутения спилловер может повысить адсорбционную способность системы, степень и скорость восстановления носителя. Это происходит за счёт диффундирования водорода с инициирующей фазы (Pt) на фазу, выполняющую роль акцептора (оксидный носитель), и, тем самым, осложняет фиксирование конечной точки осаждения и отсечение фарадеевского и емкостного токов. Аналогичные проблемы возникают и из-за спилловера кислорода, но проявляются уже в области больших потенциалов (более 0,5 В) и вносит погрешность в метод определения удельной поверхности Pt по десорбции CO. Стоит отметить что спилловер вносит меньшую погрешность в измерения, проведенные по методике, основанной на десорбции адатомов меди, чем в аналогичных методиках измерения по водороду и монооксиду углерода.
2.2.2. Растворение оксидов
При определении площади удельной поверхности платины по H2 области потенциалов десорбции и восстановления оксидных носителей платины могут пересекаться, что повлечет за собой изменение емкостных токов, окажет значительное негативное влияние на точность измерений и сделает образец не пригодным к использованию.
Кулонометрический метод измерения поверхности платины, основанный на интегрировании пика десорбции CO на вольтамерограмме так же осложняется при использовании оксидных носителей платины. Процесс адсорбции монооксида углерода на композитный материал может сопровождаться восстановлением оксидного носителя. Из-за этого на пик десорбции монооксида углерода накладываются токи, связанные с окислением восстановленной фазы. Разделить вклады этих двух, параллельно протекающих процессов, не представляется возможным[9].
2.3. Вывод.
Электрохимические способы измерения поверхности платины, нанесённой на твёрдый раствор диоксида рутения в диоксиде титана являются наиболее подходящими и распространёнными для поставленной задачи. Кулонометрические методы отличаются большой сходимостью и воспроизводимостью, но, вследствие специфики образца, имеют ряд недостатков. Явление спилловера водорода и восстановление оксида-носителя изменяют положение базовой линии на вольтамерограмме и тем самым сильно снижают точность измерения площади активной поверхности по десорбции H2 и CO. Тот факт, что емкость двойного электрического слоя не меняется при переходе от раствора серной кислоты к раствору с добавкой Cu2+, и то, что десорбция адатомов меди на платине происходит при потенциалах ≈ 0,4-0,8 В делает определение по десорбции меди наиболее точным и предпочтительным для данных образцов. Однако, поскольку заполнение поверхности платины адатомами меди зависит от потенциала осаждения, времени осаждения и концентрации меди в растворе метод требует оптимизации.
3. Методика исследований.
3.1. Подготовка рабочего электрода.
Подготовка образца к измерению производилась следующим образом: в 1 мл изопропанола, содержащего 10 мкл протонпроводящего полимера Nafion, диспергировали 10 мг 20%Pt/Ti0,92Ru0,08O2 и 1 мг углеродных нанотрубок. Обработку в ультразвуковой ванне в течении часа повторяли перед каждым нанесением.
В качестве рабочего выступал дисковый стеклоуглеродный электрод с площадью поверхности 7,07 мм2, на который последовательно три раза наносилось 0,7 мкл подготовленной суспензии.
3.2. Электрохимическая ячейка.
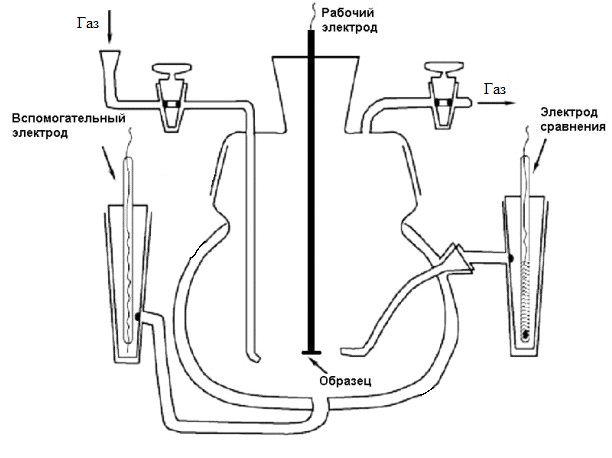
Измерения производились на приборе Elins "ПИ 50-ПРО" в трехэлектродной электрохимической ячейке, которая изображена на рис.5.
Рисунок №5. Схема трёхэлектродной ячейки.
Рабочим электролитом служил 0,5М раствор серной кислоты. При проведении измерений по десорбции меди раствор так же содержал CuSO4 с концентрацией 10мМ. В качестве электрода сравнения использовался нормальный водородный электрод (НВЭ) в растворе H2SO4 с концентрацией 0,5М. Вспомогательный электрод – платиновая фольга. В качестве газовой среды для измерений по методике десорбции монооксида углерода использовался CO, в остальных случаях – аргон. Измерения производились после полного замещения кислорода, растворённого в фоновом электролите, на рабочий газ. Удаление остаточного кислорода отслеживалось по фоновым кривым.
3.3. Режим измерений.
Для образцов снимались вольтамперограммы со скоростью развертки 10мВ/с. С целью получения пиков десорбции CO и Cu на ЦВА образцы подвергались предобработке в потенциостатическом режиме. Для монооксида углерода использовался потенциал 100 мВ (НВЭ), для меди он варьировался от 170 до 300 мВ (НВЭ). При регистрировании пика десорбции для водорода был использован диапазон потенциалов от 01.01.01 мВ (НВЭ), для монооксида углерода от 100 до 1200 мВ (НВЭ), а для меди нижний порог был равен потенциалу осаждения, верхний был постоянен – 1200 мВ (НВЭ).
4. Результаты и их обсуждение.
Все полученные циклические вольтамперограммы (рис.6-8) имели характерную форму для образцов, содержащих поликристаллическую платину. Кривые хорошо воспроизводились и легко поддавались обработке посредством интегрирования фарадеевских токов. Восстановления оксидного носителя при адсорбции CO не наблюдается.
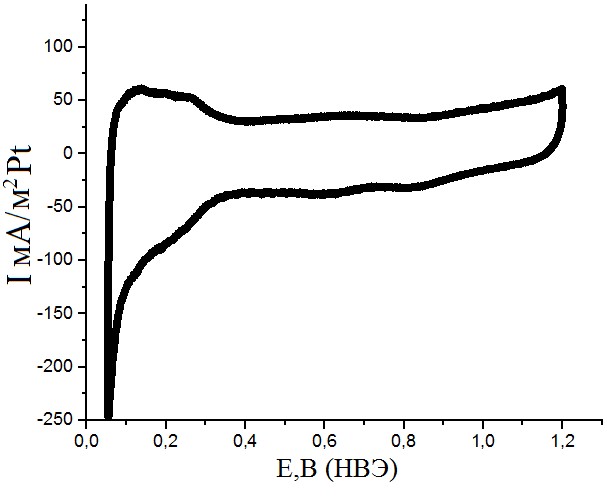
Рисунок №6. Фоновая кривая. Скорость развертки 10мВ/с. Раствор с концентрацией H2SO4 0,5М.
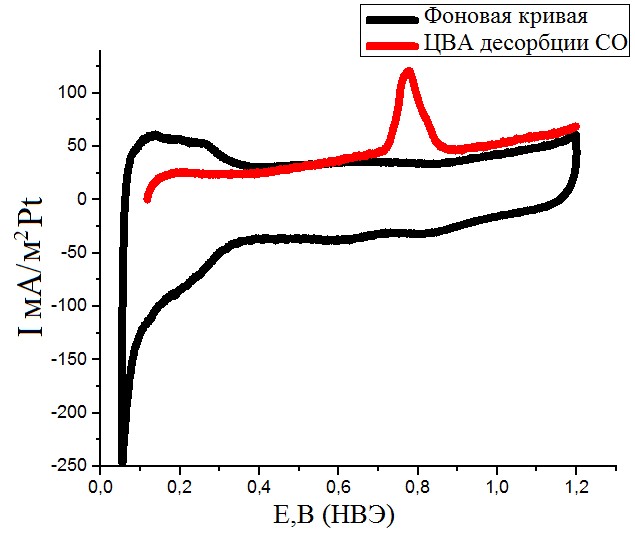
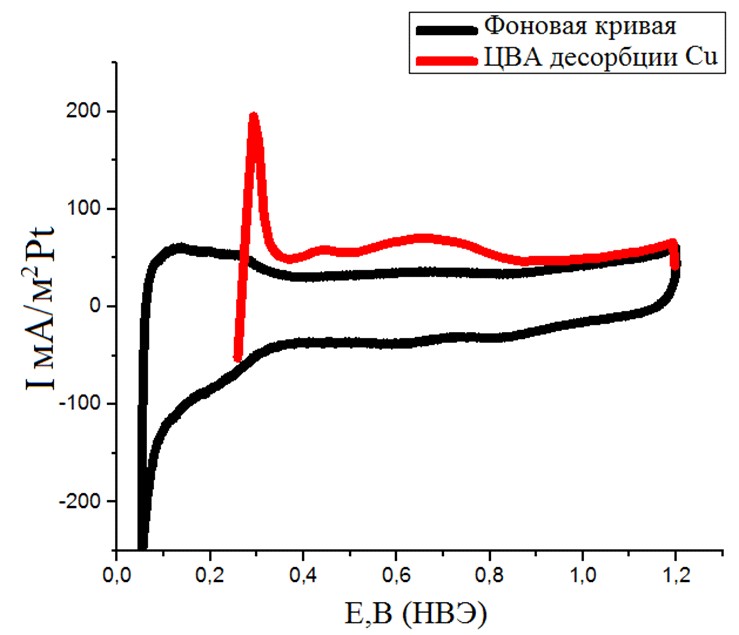
Рисунок №7. ЦВА десорбции CO. Скорость развертки 10мВ/с. Раствор с концентрацией H2SO4 0,5М. Продолжительность адсорбции – 3 минуты.
Рисунок №8. ЦВА десорбции Cu. Скорость развертки 10мВ/с. Раствор с концентрациями H2SO4 0,5М и CuSO4 0,01М. Продолжительность адсорбции – 3 минуты.
В процессе обработки результатов получена зависимость отношения площадей пиков десорбции меди и водорода на ЦВА от потенциала проведения адсорбции Cu на поверхность активного вещества при постоянном времени осаждения t = 3 мин (рис.9).
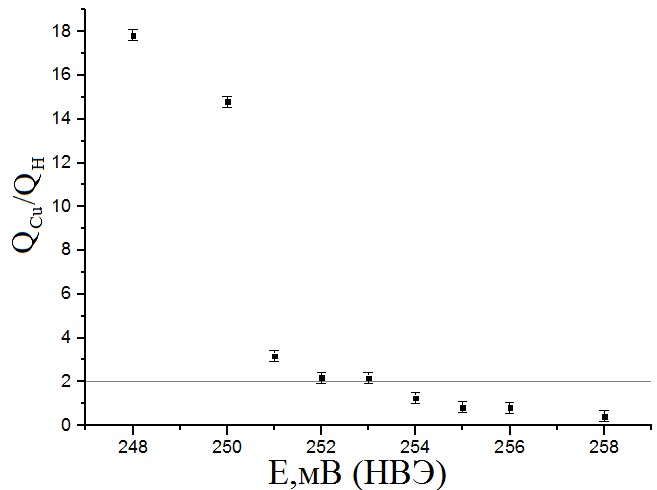
Рисунок №9. Зависимость отношения площадей пиков десорбции меди и водорода на ЦВА от потенциала адсорбции меди. Время адсорбции меди – 3 минуты. Концентрация Cu2+ 10мМ.
По полученной зависимости можно установить 3 интервала потенциалов. В области E < 252 мВ (НВЭ) результаты, соответствующие десорбции Cu, завышают результаты, полученные при десорбции водорода, за счёт образования объёмной меди. При потенциалах ≈ 252-253 мВ (НВЭ) площади пиков соотносятся как 2:1 (что соответствует отношению зарядов Cu2+ и H+). В области E > 253 мВ (НВЭ) адатомы не полностью заполняют активную поверхность. Получена зависимость отношения площадей пиков десорбции меди и водорода на ЦВА от времени, в течении которого велась адсорбция Cu на поверхность Pt при постоянном потенциале E=252 мВ (НВЭ) (рис.10).
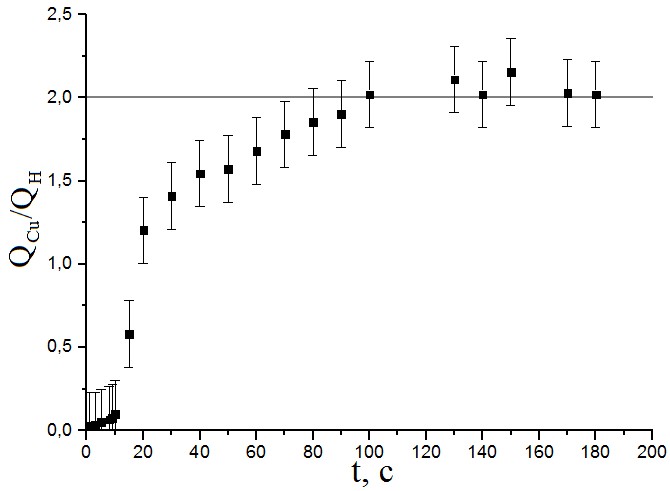
Рисунок №10. Зависимость отношения площадей пиков десорбции меди и водорода на ЦВА от длительности процесса адсорбции меди. Потенциал, при котором производилось осаждение E=252 мВ относительно НВЭ. Концентрация Cu2+ 10мМ.
Отчётливо видно, что заполнение поверхности платины адатомами меди заканчивается через 110-120 секунд потенциостатической обработки, после чего завершенный монослой не претерпевает изменений.
После калибровки метода по пику десорбции адатомов меди на ЦВА была рассчитана удельная площадь поверхности платины на оксидном носителе, которая составила 18±2 м2 на 1 грамм Pt. Это значение, с учётом погрешности, согласуется со значением 16±2 м2/г, полученным при обработке пика десорбции CO.
