

Партнерка на США и Канаду по недвижимости, выплаты в крипто
- 30% recurring commission
- Выплаты в USDT
- Вывод каждую неделю
- Комиссия до 5 лет за каждого referral
УДК 621.3.038.825.7; 530.182
В. Буртман*,**, *, Милко Ван дер Бум***, *, *, *, Е. н. Никонова*
Формирование органических тонких пленок нелинейных оптических материалов методом молекулярной послойной эпитаксии2
Описаны условия формирования пленок [(аминофенил)азо]пиридина методом молекулярной послойной эпитаксии, проведено исследование их оптического поглощения и рентгеновской фотоэлектронной спектроскопии. Нелинейные свойства таких структур описаны при помощи измерения интенсивности генерации второй гармоники (ГВГ) от угла падения.
Ключевые слова: молекулярная послойная эпитаксия, генерация второй гармоники
Введение
Создание материалов для нелинейной оптики с приемлемыми оптическими, химическими и термическими свойствами представляет большой интерес [i,ii,iii,iv,v,vi,vii,viii,ix]. Так, методы Ленгмюра-Блоджетт и электростатической самосборки широко используются для получения органических тонких пленок (моно - и многослойных) с эффективными нелинейными оптическими (НЛО) свойствами, в то время как методы центрифугирования и химического осаждения из газовой фазы применяются гораздо реже [x,xi,xii,xiii,xiv,xv]. Известно, что моно - и многослойные структуры [(аминофенил)азо]пиридин-производных со значительными НЛО свойствами могут быть получены методом центрифугирования с последующей термообработкой в вакуумной печи, либо относительно медленными методами осаждения из раствора [2]. Наиболее сложным является создание НЛО материалов с ковалентно связанными органическими молекулами на поверхности кремния, перспективных для создания оптоэлектронных устройств [xvi].
В работе рассмотрена возможность использования молекулярно-послойной эпитаксии (МПЭ) для создания НЛО тонкопленочных структур из газовой фазы. Этот подход может быть использован для изготовления эффективных органических светоизлучающих диодов (ОСИД) и органических структур, проявляющих свойства многослойных конденсаторов [xvii].
Эксперимент
Все операции, связанные с очисткой и подготовкой подложек выполнялись в атмосфере сухого азота. Для получения НЛО пленок очищенные стеклянные подложки погружались в обезвоженный раствор гептана и фенилтрихлорсилана (объемное соотношение 100:1) при комнатной температуре в течение 20 мин. Затем подложка промывалась обезвоженным гексаном, обрабатывалась ультразвуком в ацетоне в течение 1 мин [ 1,2,xviii,xix,xx]. В результате проделанных операций на поверхности подложки формировался плотноупакованный мономолекулярный слой бензилхлорида (1). Затем эта подложка устанавливалась в ламинарный реактор [13,17] для формирования на ее химически модифицированной поверхности тонкопленочной структуры соединения 3 (рис.1) методом химической адсорбции. Для этого подложка в реакторе нагревалась до T = 178 °C, после чего на нее наносился хромофор 4-[[4-[N, N-бисгидроэтил)аминофенил]азо]пиридин (2), при температуре испарения T = 150 °C в течение 25 мин при общем давлении 50 мТорр. При этом выполнялись условия получения ламинарного потока пара реагента с числом Рейнольдса Re порядка 100. В результате реакции нуклеофильного замещения по схеме, приведенной на рис. 1, на поверхности образовывался ковалентно связанный двойной слой (3). После осаждения образцы медленно охлаждались в реакционной камере в потоке аргона. Отметим, что подобный способ формирования пленок с кватернизацией хромофоров хинолина может использоваться для модификации электронных свойств поверхности кремния [20].
Следущие приборы использовались для изучение самособирающихся структур: (1) X-ray Photoelectron Spectrometer (XPS) модели Kratos AXIS Ultra, (2) эллипсометр variable angle spectroscopic ellipsometer (Woollam Co.) VASE 32 PC и (3) спектрофотометер Shimadzu UV-Vis-NIR 3101 PC scanning spectrophotometer.
Рис. 1. Схема молекулярной сборки пленки 3 по методу МПЭ
Полученная пленка 3 является термостойкой и инертной к большинству органических растворителей, а ее формирование однозначно идентифицируется методами электронной спектроскопии, эллипсометрии, рентгеновской фотоэлектронной спектроскопии (XPS) и угловой зависимости генерации второй гармоники (ГВГ).
Результаты и их обсуждение
В спектре поглощения пленки 3 (рис.2а) максимум поглощения находится на 580 нм и смещен в красную область по сравнению с начальным хромофором 2, у которого максимум поглощения приходится на 443 нм в метаноле), что говорит об образовании комплекса с переносом заряда и доказывает взаимодействие пиридильной части с бензилхлоридом. Известно, что соединение 2 легко реагирует с алкилгалогенидами в растворах, например, реакция соединения 2 с избытком метилйодида в тетрагидрофуране дает йодидную соль 4-[[4-[бис(гидроксиэтил)амино]фенил]азо]-1-метилпиридина пурпурного цвета. При этом, в результате переноса заряда, наблюдается такой же сдвиг максимума поглощения, как и в поверхностно-связанном хромофоре 3.
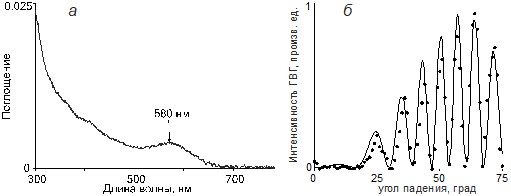
Рис. 2. Спектр электронного поглощения (а) и зависимость интенсивности ГВГ от угла падения пленки 3
Эллипсометрические измерения показали, что толщина изготовленной пленки 3 составляет ~21 Е, что хорошо согласуется с ранее выполненными исследованиями [1,2]. Спектр XPS пленки 3 показал, что на поверхности находится примерно равные количества ковалентно связанного хлора (199.8 эВ, 13.0 %), а также его иона Cl - (197.7 эВ, 17.8 %). Сдвиг в 2.1 эВ связан с меньшей энергией связи при образовании Cl - [2,xxi]. Наличие соединения 3 на поверхности подложки также подтверждается сигналом N 1s на 397.8 эВ с интенсивностью 69.2 %. Отметим, что критериями протекания реакции по схеме, приведенной на рис.1, являются величины отношений интенсивностей N/Cl - и N/Cобщее и по теоретическим оценкам при полном протекании реакции они должны быть равны 4. В нашем случае в спектре XPS отношение N/Cl‑=3.8, а N/Cобщее=2.2, что позволяет говорить о том, что в ходе реакции МПЭ ~55 % бензилхролидных фрагментов 1 подверглись реакции с соединением 2 с образованием пленки 3.
Предыдущие исследования показали [1-3], что, зависимости интенсивности ГВГ от угла измерения позволяют идентифицировать и определять характеристики самоорганизующихся пленок класса [(аминофенил)азо]пиридина, причем как монослойных, так и многослойных. Измерения интенсивности поляризованной ГВГ были выполнены на установке, описанной в работе [3], на длине волны 1064 нм с модуляцией добротности. Зависимость интенсивности ГВГ от угла падения, приведенная на рис. 1б показывает, что качество и однородность органической пленки на обеих сторонах подложки идентичны. Эта зависимость может быть описана уравнением (1), где Ш – угол синхронизма.
![]() (1)
(1)
Объемный отклик второго порядка нелинейной восприимчивости ч(2)zzz ~ 7Ч10-8 СГСЭ и угол синхронизма Ш ~ 36 ° для молекул хромофора 3 получен путем калибровки данных ГВГ относительно кварца. Этот угол близок к углу синхронизма пленки 2 и ее производных, полученных другими методами [1-3], для которых Ш ~ 38-42 °. Нелинейная восприимчивость для пленок, полученных в газовой фазе ниже, чем у аналогичных пленок 2, полученных методами центрифугирования или выращенных в растворах (ч(2)zzz ~ 5Ч10-7 СГСЭ). Однако объемный отклик ч(2)zzz пленки 3 сравним с откликом соединений 4-[N, N-бис(три-гидроксипропил)амино]фенилэтил-4'-пиридина, для которого ч(2)zzz=6Ч10-8 СГСЭ и 4-транс-4(4-(10-фенотиазинил)бутирамидо)стирил)пиридина (ч(2)zzz=2.8Ч10‑8 СГСЭ) [7,8].
Заключение
Представлен способ создания тонких органических пленок, где хромофор ковалентно присоединен к химически модифицированной поверхности подложки по реакции нуклеофильного замещения. Показано, что созданные таким способом пленки обладают НЛО свойствами. Несмотря на то, что реакция самосборки не была оптимизирована и выход составил всего 55 %, установлено, что объемный отклик нелинейной восприимвичости ч(2)zzz ~ 7Ч10-8 СГСЭ. Таким образом, метод молекулярно-послойной эпитаксии позволяет формировать органические тонкопленочные материалы с нелинейными оптическими свойствами.
Список литературы
*Сибирский физико-технический институт им.
Национального исследовательского Томского государственного университета,
г. Томск, Россия
**Университет Штата Юта
г. Солт-Лейк-Сити, США
*** Институт Вейцмана, Реховот, Израиль.
E-mail: *****@***utah. edu vlad. *****@***edu, *****@***tsu. ru, milko. *****@***ac. il
Буртман Владимир, PhD, профессор
, д. ф.-м. н., профессор.
Милко Ван дер Бум, PhD, декан факультета химии.
, к. х.н., ст. науч. сотр.
, к. ф.-м. н., доцент, ст. науч. сотр.
, к. ф.-м. н., науч. сотр.
, мл. науч. сотр.
1 Работа выполнена в рамках Государственного задания № 16.578.2014/К Минобрнауки России
2 Работа выполнена в рамках Государственного задания № 16.578.2014/К Минобрнауки России
i Yitzchaik S. and Marks T. J. // Acc. of Chem. Res. – 1996. – V. 29. – P. 197-202.
ii Lin W., Lin W., Wong G. K. and Marks T. J. // J. Am. Chem. Soc. – 1996. – V. 118. – P. 8034-8042.
iii Yitzchaik S., Roscoe S. B., et al. // J. Phys. Chem. – 1993. – V. 97. – P. 6958-6960.
iv Katz H. E., Wilson W. L. and Scheller G. // J. Am. Chem. Soc. – 1994. – V. 116. – P. 6636-6640
v Wang F., Harper A. W., et al. // Chem. Mater. – 1999. – V. 11(9). – P. 2285.
vi Wijekoon W. M.E. P., Wijaya S. K., et al. // J. Am. Chem. Soc. – 1996. – V. 118. – P. 4480-4483.
vii Hung W., Helvenston M. et al. // Langmuir. – 1999. – V. 15. – P. 6510.
viii Smilowitz L., Jia Q. X. et al. // J. Appl. Phys. – 1997. – V. 81 (5) – P. 2051-2054.
ix Mayer G. V., Kopylova T. N., et al. //Russian Physics Journal. – 2009. – V. 52., № 6. – P. 640-645.
x Johal M. S., Cao Y. W. et al. // Chem. Mater. – 1999. – V. 11. – P. 1962-1965.
xi Koloski T. S., Dulcey C. S., Haralson Q. J. and Calvert J. M. // Langmuir. – 1994. – V.10. – P. 3122-3133.
xii Haskal E. I., Zhang Y., Burrows P. E. and Forrest S. R. // Chem. Phys. Lett. – 1994. – V. 219. – P. 325-330.
xiii Imanishi H. S., Kakuta A. and Numata S. // Phys. Rev. Lett. – 1993. – V. 71. – P. 2098-2101.
xiv Tsuzuki T., Hirota N., Noma N. and Shirota Y. // Thin Solid Films. – 1996. – V. 273. – P. 177-180.
xv Yoshimura T., Tastuura S., et al. // Appl. Phys. Lett. – 1991. – V. 59. – P. 482-484.
xvi Ashwell G. J. Molecular Electronics, John Wiley & Sons Inc. 1992.
xvii Burtman V., Zelichenok A. and Yitzchaik Angew // Chem. Int. Ed. – 1999. – V. 38. – P. 2041-2045.
xviii Moaz R. and Sagiv J. // Langmuir. – 1987. – V. 3. – P. 1034-1044.
xix Wasserman S. R., Tao Y.-T. and Whitesides G. M. // Langmuir. – 1989. – V. 5. – P. 1074-1087.
xx Cohen R., Zenou N., Cahen D. and Yitzchaik S. // Chem. Phys. Lett. – 1997. – V. 279. – P. 270-274.
xxi Moulder J. F., Stickle W. F., Sobol P. E. and Bomben K. D. Handbook of X-ray Photoelectron Spectroscopy; Perkin Elmer: Eden Prairie, MN, 1992.
