


Партнерка на США и Канаду по недвижимости, выплаты в крипто
- 30% recurring commission
- Выплаты в USDT
- Вывод каждую неделю
- Комиссия до 5 лет за каждого referral
Унифицированный репозиторий QSAR моделей и молекул как система управления конфигурациями траекторий поиска зависимостей
«структура-свойство»
1, 1, 1, 1, 1
1Кафедра вычислительной математики, Механико-математический факультет, Московский Государственный Университет им. ,
Москва, 119991 Россия; E-mail: *****@***ru, *****@***ru
Показано, что использование моделей «структура-свойство» для массового прогнозирования свойств молекулярных структур приводит к необходимости иметь унифицированное хранение параметров преобразований молекул и их промежуточных результатов. Введено понятие «траектории построения» матрицы «Молекула-Дескриптор» (МД-матрицы), где каждая траектория определяется собственной конфигурацией. Унифицированное хранение траекторий и их конфигураций позволяет обеспечивать повторяемость результатов при прогнозировании свойств новых молекул.
Жизненный цикл QSAR моделей (Quantitative Structure Activity Relationship) имеет итерационный характер, в котором участвуют как математики, так и химики. Их интересы существенно различны: математик стремиться получить простую и прогностично устойчивую модель, но для химика такая модель – только инструмент облегчающий поиск новых химических структур с заданным значением целевого свойста. Синтез химиком веществ по результатам прогноза, дальнейшее определение их свойств позволяет на практике оценить качество QSAR модели, а затем и модифицировать ее, добавив данные о вновь синтезированных структурах в обучающую выборку.
Участие двух разных ролей в работе с QSAR моделями – математика-разработчика и химика пользователя (Рис.1), - требует «отделения» математика от построенной им модели, использующей последовательность преобразований молекул. Пользователь-химик должен суметь запустить прогноз новых молекул без присутствия математика спустя месяцы и годы после построения модели. Проблема состоит в том, что последовательность преобразований может быть построена на основе модулей и программ, имеющих различную алгоритмы и различные значения входных параметров. Так, например, в интернете можно найти
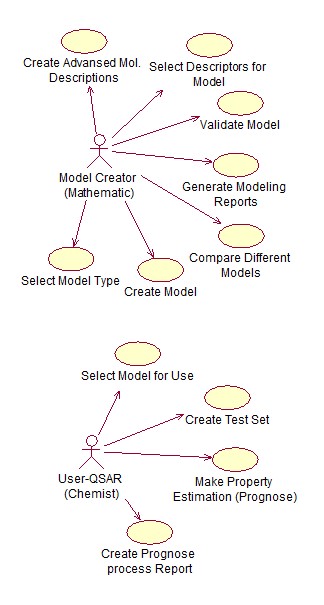
Рис. 1. Диаграмма UML, представляющая основных пользователей системы и их сценарии использования
различные программы, позволяющие построить пространственную укладку атомов молекулы, т. е вычислить ее «3D-конформацию». Эти программы используют различные принципы, начиная с методов молекулярной механики и кончая квантово-химическими расчетами на основе уравнения Шредингера и специальных версий силовых полей.
При проведении прогнозировании свойств новых молекул химиком, новые структуры должны пройти тот же путь преобразований теми же самыми программными модулями, что прошли молекулы обучающей выборки при построении модели математиком. Это означает, что все версии программ, (участвующие в цепочке преобразований молекул) и их параметры должны единообразно сохраняться в некотором репозитории, чтобы была возможность полной повторяемости результатов модели при прогнозировании.
Молекулярные структуры после построения QSAR модели не должны «стираться», поскольку, во-первых, они определяют «область допустимых значений» (ОДЗ) модели и, во-вторых, могут быть повторно использованы (при появлении новых данных о механизмах действия) для QSAR моделирования.
Постановка задачи
При формировании строк матрицы «Молекула-Дескриптор» (МД-матрицы) для каждой молекулы нужно провести:
1. Расчет устойчивых пространственных конформаций;
2. Построение молекулярной поверхности (в виде триангулированной поверхности);
3. Расчет потенциала на точках поверхности (локальное физ.-хим. свойство) [1];
4. Идентификация особых точек (ОТ) на поверхности [2];
5. Классификация ОТ (по геометрии и по физико-химическому свойству) [3];
6. Полное перечисление всех k-фрагментов (k = 2, 3, 4, ... ) [4];
7. Символьное кодирование k-фрагментов – (имя дескриптора = код фрагмента) [52].
Формирование матрицы проводится путем объединения списка дескрипторов, вычисленных по каждой молекуле обучающей выборки.
Назовем траекторией построения МД-матрицы промежуточные данные-результаты, получаемые в ходе выполнения шагов 1-7. Назовем конфигурацией траектории набор версий программных модулей и их входных параметров, на основе которых выполняется цепочка шагов 1-7.
Целью создания репозитория (системы) является информационная поддержка хранения, сравнения и использования (для поиска новых химических структур) ранее построенных QSAR моделей – независимо от времени их создания, - на основе их единого представления в реляционной СУБД. Система должна сопровождать QSAR модели, построенных на различных дескрипторах молекул.
Наша задача состояла в том, чтобы спроектировать необходимый состав элементов репозитория и построить модели «сущность–связь» для траекторий (и их конфигураций) на основе реляционной модели базы данных. Построение было проведено с использованием визуальной нотации UML (Unified Modeling Language) [6].
На рисунке 2 показана модель «сущность-связь» (в форме диаграммы классов UML) для траектории построения МД-матрицы до уровня дескрипторов 2-фрагментов. На рисунках 3 и 4 показаны сущности для хранения элементов и конфигураций различных «отрезков» траектории создания МД-матрицы.
Аналогично, можно определить траекторию построения зависимости «структура-свойство». Такая зависимость может быть получена на основе разных базовых моделей [7].

Рис. 2. Диаграмма классов UML, представляющая структуру хранения траектории построения МД-матрицы до уровня 2-фрагментов
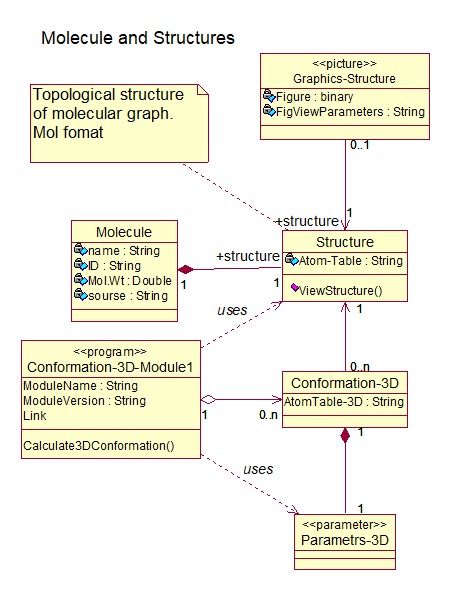
Рис. 3. Диаграмма UML, представляющая элементы хранения «начала» траектории совместно с элементами конфигурации – программным модулем и параметрами расчета 3D конформаций
Так, если за основу QSAR модели выбрать кусочно-линейную зависимость, то цепочка ее построения будет состоять из следующих задач:
1. Анализ значимости дескрипторов, объединение/отброс столбцов МД-матрицы.
2. Проведение линейного разведочного анализа:
2.1. Построение линейной модели (на всей выборки) на основе метода группового учета аргументов (МГУА)[2];
2.2. Построение «области допустимых значений» (ОДЗ) модели в виде условий, позволяющих отказаться от прогнозирования свойства «чужой» молекулы;
2.3. Проверка устойчивости модели с использованием скользящего контроля;
2.5. Принятие решение о необходимости построения кластерной структуры обучающей выборки (если проверка на выявила неустойчивость или низкое качество линейной апроксимации).
3. Проведение кластерного анализа и поиск линейной зависимости на каждом кластере:
3.1. Выбор вида метрики и дескрипторов для ее вычисления по маске (Рис. 5);
3.2. Выбор алгоритма кластерного анализа и построение кластерного разбиения обучающей выборки;
3.3. Построение линейной модели на кластерах. Для каждого кластера (K):
K.1. Формирование МД-матрицы по структурам, вошедшим к данный кластер
K.2. Построение линейной модели (на выборке кластера);
K.3. Построение «нечеткой» (fuzzy) модели прогнза (на выборке кластера);
4. Верификация (тестирование) построенной кусочно-линейной модели (скользящий контроль на выборках кластера).
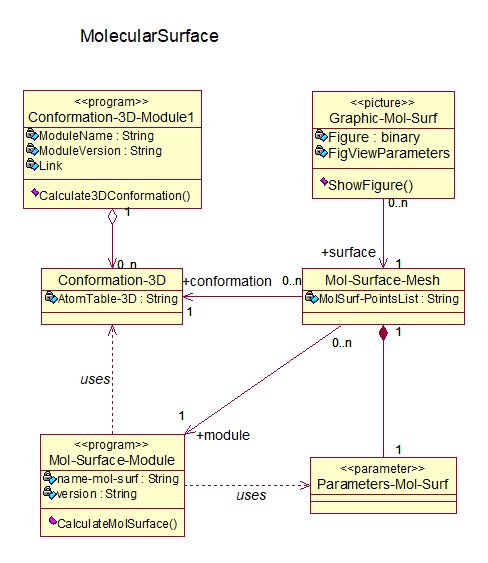
Рис. 4. Элементы хранения данных при построении участка траектории вычисления молекулярных поверхностей.
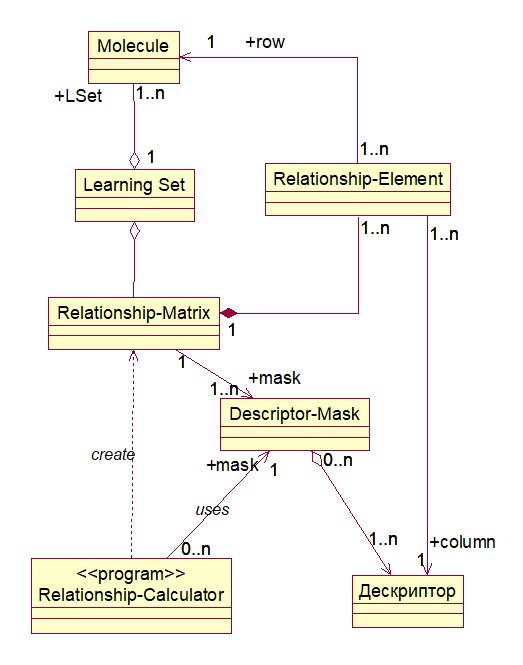
Рис. 5. Диаграмма классов UML, определяющая структуру данных при построении матрицы расстояний (отношений) между молекулами для кластерного анализа.
Заключение
Необходимость «отделения» QSAR моделей от их разработчиков, приводит к необходимости иметь систему-репозиторий, в которой проводилось структурированное модели «структура-свойства» храняться и сопровождаются на протяжении их жизненного цикла.
Введено понятие траектории и конфигурации процесса построения МД-матрицы (Молекула-Дескриптор), для них построены модели «сущность-связь». Это позволяет приступить к физическому проектированию структуры реляционной базы данных, лежащей в основе репозитория.
