
Партнерка на США и Канаду по недвижимости, выплаты в крипто
- 30% recurring commission
- Выплаты в USDT
- Вывод каждую неделю
- Комиссия до 5 лет за каждого referral
Публикация доступна для обсуждения в интернет как материал “Всероссийской рабочей
химической конференции “Бутлеровское наследие-2011”. http:///bh-2011/
Поступила в редакцию 4 февраля 2011 г. УДК 544.18, 544.431.133.
Тематическое направление: Квантово-химический расчет некоторых элементарных актов реакций кислотно-каталитического разложения гидропероксида кумола. Часть 1.
Протонизация гидропероксида кумола
© *+ и
Кафедра общей химической технологии. Казанский государственный технологический университет.
Ул. К. Маркса, 68. г. Казань, 420015. Республика Татарстан. Россия.
Тел.: (843) 231-42-52. E-mail: *****@***ru
___________________________________________
*Ведущий направление; +Поддерживающий переписку
Ключевые слова: квантово-химический расчет, гидропероксид кумола, кислотно-каталитическое разложение, фенол, ацетон, диметилфенилкарбинол, α-метилстирол.
Аннотация
Кислотно-каталитическое разложение гидропероксида кумола протекает с высокой скоростью через последовательное образование промежуточных интермедиатов, детектирование которых физи-ческими методами затруднено. Проведение квантово-химического исследования данного процесса позволило установить наличие различных устойчивых конформаций гидропероксида кумола, отличающиеся механизмами их дальнейшего превращения. В работе предложена возможность протонизации по обоим атомам (алкоксильного и гидроксильного) кислорода гидропероксида кумола разных конформаций. Найдено, что при протонизации гидроксильного кислорода гидропероксида кумола дегидратация последнего сопровождается синхронной перегруппировкой иона оксония в ион карбония, который далее взаимодействует со второй молекулой гидропероксида с образованием промежуточного комплекса, разлагающегося в фенол, ацетон и ион карбония. В случае протонизации алкоксильного атома кислорода отщепляется пероксид водорода с образованием карбокатиона, который превращается в α-метилстирол или в диметилфенилкарбинол.
Введение
Гетеролитическое разложение гидропероксида кумола (ГПК) в присутствии кислоты было открыто в 1942 году советским химиком Удрисом Рудольфом Юрьевичем (1899-1949), что заложило основу нового промышленного способа производства фенола. Получение ГПК и его кислотно-каталитическое разложение составляют основу кумольной технологии совмест-ного производства фенола и ацетона [1]. Реакция получения фенола и ацетона из ГПК протекает с высокой скоростью и выделением большого количества теплоты, что несколько затрудняет контролировать процесс и исследовать протекающие сложные реакции в системе. Квантово-химические расчеты являются удобным инструментом проведение исследования данного процесса.
В присутствии небольшого количества сильной кислоты ГПК распадается с образо-ванием фенола и ацетона. Реакция протекает по сложному механизму ионного типа с проме-жуточным возникновением положительно заряженных ионов. Механизм кислотно-каталити-ческого разложения ГПК предложенный Харашем [2] и подтвержденный последующими ис-следователями [3-7] выглядит следующим образом:
Схема 1
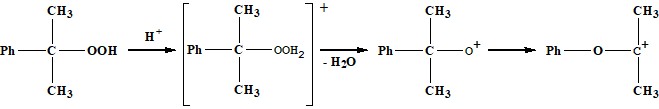
Схема 2
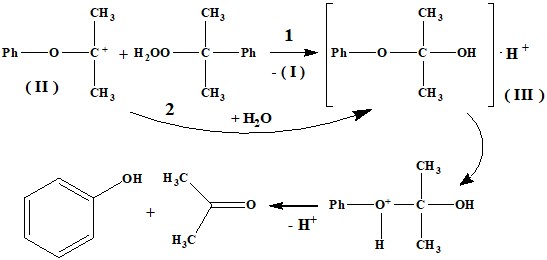
Основываясь на кинетических данных, Хараш предположил, что образующийся карбо-катион (II) затем взаимодействует со второй молекулой гидропероксида. Авторами работы [3] было предложено, что карбокатион (II) может взаимодействовать с молекулой воды с образованием комплекса (III). Однако, по мнению авторов [6] если направление 2 имеет место быть, то его вклад очень маленький, основной путь превращения карбокатиона (II) является направление 1, который хорошо согласуется экспериментальными данными.
Исследование кислотно-каталитического разложения ГПК в водной среде, содержащей Н2О18 [6] показало, что в феноле отсутствует атом О18, из чего был сделан вывод – вода не принимает участие в процессе, как показано в схеме 2. Попытка определить содержания О18 в ацетоне у авторов не увенчался успехом. Из схемы 2 вытекает, что атом О18 более вероятно мог остаться в молекуле второго продукта – в ацетоне, судя по всему кислородный атом гидроксильной группы комплекса (III) не мигрирует. Следовательно, вопрос об участии воды в превращении ГПК остается открытым.
Анализ опубликованных работ показывает, что мнение исследователей относительно механизма кислотно-каталитического разложения гидропероксидов совпадают в принципе, отличие имеется только в деталях.
Имеются все основания считать, что энергия сольватации комплекса SH+ гидроперок-сидом невелика, так как координационные вакансии протона уже в значительной степени насыщены [8]. Поэтому наиболее существенный вклад в величину энергии пересольватации вносит энергия взаимодействия между растворителем и гидропероксидом. Исследование кинетики разложения ГПК в ацетоне в присутствии гетерополикислот [9] показало, что в процессе имеет место общий кислотный катализ, перенос протона к молекуле ГПК является медленной стадией. К такому же выводу пришли авторы работ [10], протонированный ГПК может быть короткоживущим интермедиатом или переходным состоянием реакции, струк-тура которого показано ниже:
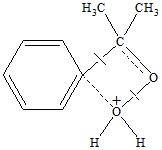
и [11] ссылаясь на то, что указанный интермедиат не удается наблюдать даже при -60 oС [12] пришли к мнению о том, что процесс идет в одну стадию; протонизация ГПК сопровождается синхронной перегруппировкой в конечные продукты. Таким образом, для гетеролитического разложения необходимость протонизации ГПК авторами не отрицается.
Несогласие с приведенной выше схемой Хараша в более яркой форме выражает Зако-шанской [13-15] по мнению, которого передача протона от кислотной частицы к молекуле ГПК не происходит, и реакция протекает не в рамках карбоний-ионного механизма, а за счет межмолекулярной (кислотная частица-субстрат) и внутримолекулярной перегруппировок в молекуле ГПК под влиянием атакующих кислотных частиц (схема 3).
Схема 3

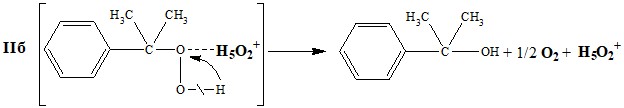
*– в оригинале была допущена опечатка (в схеме вместе ГПК нарисовано ДМФК),
АН – молекула кислоты
Необходимо отметить, что в водной среде заметное разложение ГПК происходит только в присутствии больших концентраций сильных кислот (15-40%-ной H2SO4), хотя кислотные частицы, в том числе Н5О2+ существуют и при малых концентрациях кислоты, однако расходования гидропероксида при этом практически не происходит.
Изучение влияния растворителей на процесс кислотно-каталитического разложения ГПК [16] показало, что в присутствии каталитических количествах серной кислоты и темпе-ратуре 50 oС заметное разложение ГПК в воде, этаноле, диметилсульфоксиде, формамиде также не происходит.
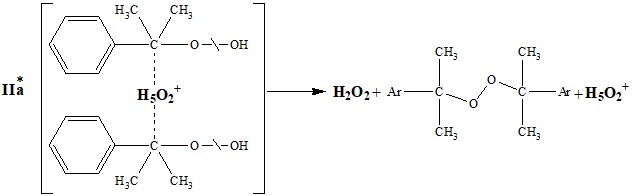
Образование комплекса (I) и внутримолекулярный механизм перегруппировки был предложен еще авторами работы [10]. Предложенная Закошанским схема участия иона Н5О2+ по реакциям IIa и IIб усложняет понимания механизма. Однако им, совместно с авторами работ [14, 17] было сделано интересное предположение, что разложение ГПК на гетеро-литические (фенол и ацетон) и на продукты гомолитического распада осуществляется через комплексы ГПК с катализатором по разным атомам кислорода пероксидной группы. Это предположение имеет значение в понимании особенности кислотно-каталитического разло-жения ГПК и может быть использовано для установления механизма реакции в различных условиях процесса (природы растворителей и добавок, в том числе катализаторов).
Исследования показывают, что если в реакции кислотного разложения использовать чистый ГПК и процесс вести при температурах, когда термическим распадом ГПК можно пренебречь, то полного превращения гидропероксида на фенол и ацетон не происходит. В растворе реакционной массы обнаруживаются и побочные продукты разложения ГПК, такие как диметилфенилкарбинол (ДМФК), пероксид дикумила (ПДК), ацетофенон, α-метилстирол (МС), т. е. продукты, так называемые гомолитического распада ГПК. Наличие радикального распада гидропероксида в присутствии кислотных катализаторов экспериментально было доказано Соляниковым [18, 19] с помощью реакции окисления стирола и изопропилового спирта под действием кислорода, выделяющегося из гидропероксида.
Доля продуктов гомолитического распада ГПК зависит от параметров реакционной среды [16], растворитель принимает участие почти во всех стадиях этого сложного процесса. Этот факт дает надежду на возможность управление процессом каталитического разложения ГПК и повышение его селективности путем подбора растворителей.
Таким образом, необходимо уточнить механизм кислотно-каталитического разложения ГПК и роль растворителей в данном процессе. Если удастся установить взаимосвязь между кинетикой равновесного перехода конформаций ГПК с участием растворителя (или другого агента), кинетику взаимодействия катализаторов с различными конформациями ГПК или интермедиатами его превращения, то можно сформулировать предсказательную базу выбора растворителя для проведения конкретного процесса с участием гидропероксидов, в частности ГПК.
Использованием квантово-химического расчета нами проведено моделирование элемен-тарных актов реакции кислотно-каталитического разложения ГПК до фенола и ацетона. Результаты первого этапа исследования, а именно протонизация молекул ГПК различными кислотными частицами, проводится ниже.
Результаты и их обсуждение
Для пояснения эффектов растворителей нами было проведено квантово-химическое исследование некоторых стадий кислотно-каталитического разложения ГПК. Вначале была проведена оптимизация структуры гидропероксида кумола. Найдено, что неассоциированная мономерная форма ГПК имеет несколько конформаций, получаемых при вращении вокруг связей С–О и О–О. Энергетически наиболее выгодной является конфигурация ГПК (рис. 1а), имеющая молекулярную водородную связь типа Н…π, образующуюся между атомом гидро-пероксидного водорода и π-электронами бензольного ядра. Однако почти во всех рассмот-ренных нами реакциях участвует другая конформация ГПК, в которой гидропероксидный фрагмент (O–O–H) развернут в противоположную сторону относительно бензольного кольца (рис. 1б). Конформация (б) энергетически несколько менее выгодна (на 7.6 кДж/моль), но в силу отсутствия дополнительной стабилизации фрагмента O-O-H (как в случае конформации (а) более реакционноспособна.
|
|
а) Cum-O1(-0.31) - O2(-0.41) - H(+0.41) | б) Cum-O1(-0.33) - O2(-0.41) - H(+0.41) |
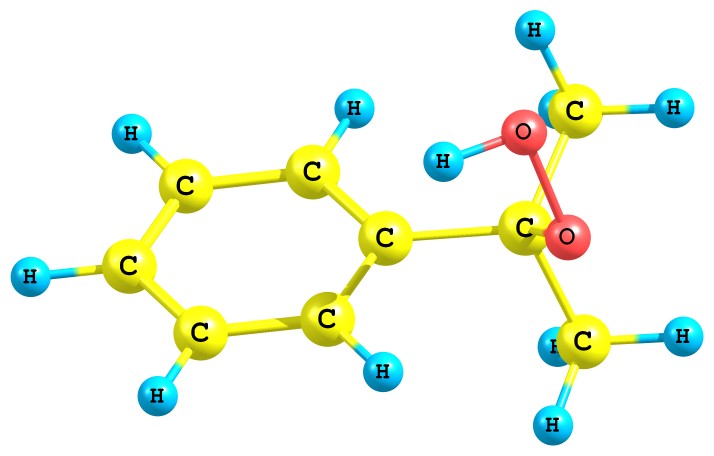
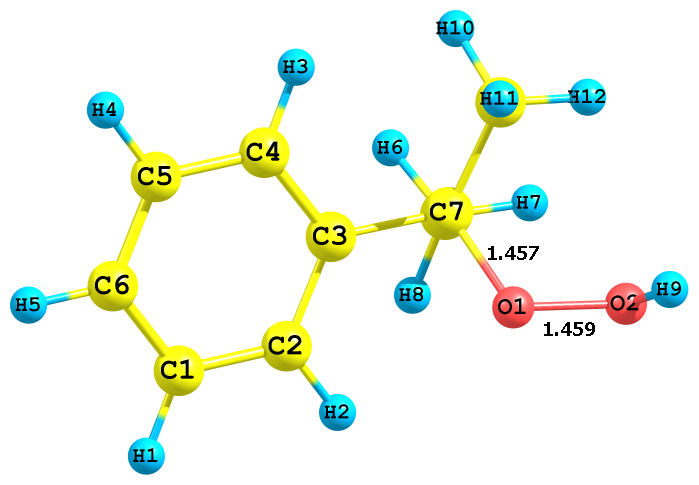
Рис. 1. Стабильные конформации молекулы ГПК
Как видно из схемы 1 для гетеролитического превращения гидропероксида необходимо его протонизация. Поскольку свободная частица H+ в растворе не существует, рассматривался вариант участия в реакции протонизации молекулы ГПК иона гидроксония H3O+ и протона, сольватированного другими растворителями. Нам не удалось локализовать переходное состояние реакции присоединения иона гидроксония H3O+ к молекуле ГПК ни путем прямого поиска, ни путем сканирования поверхности потенциальной энергии по направлению данной реакции. При приближении иона гидроксония к молекуле ГПК происходят дальнейшие превращения. Это обстоятельство говорит о том, что данная реакция протекает либо совсем безактивационно, либо с очень малой энергией активации.
На рис. 1 приведены также заряды на атомах реакционного центра для обоих конфор-маций. По абсолютным значениям заряды на атомах кислорода гидропероксида очень близки и, следовательно, предпочтительность протонизация того или иного атома кислорода будет определяться другими факторами, например стерическим эффектом. Представляет как теоре-тический, так и практический интерес исследования разложения ГПК с активацией различных атомов кислорода.
В зависимости от того, какой из гидропероксидных кислородов, алкоксильный или гидроксильный (Cum–O1–O2–H), подвергается протонизации, продукты реакции оказываются различными. Так, в случае атаки иона H3O+ по O1 происходит образование иона Ph–C+(CH2)2 (I) и молекул H2O2 и H2O (рис. 2а). Далее карбокатион (I) превращается в продукты, как показано в схеме 5. В случае же атаки H3O+ по O2 происходит образование катиона Ph–O–C+(CH3)2 (II) и двух молекул воды (рис. 2б).
|
|
а) ДHр = -120.2 кДж/моль, ДGр = -175.5 кДж/моль | б) ДHр = -370.9 кДж/моль, ДGр = -420.3 кДж/моль |
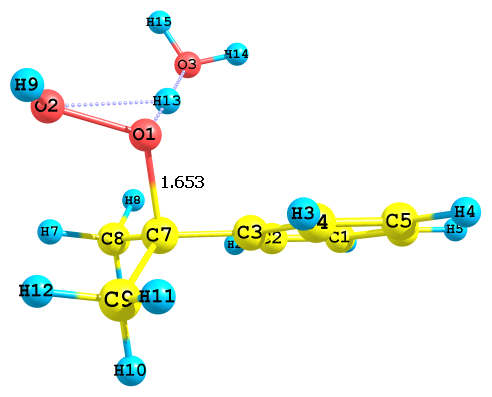
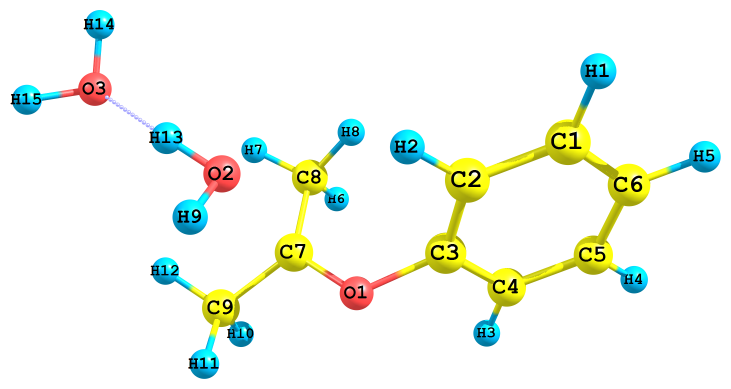
Рис. 2. Результат протонизации ГПК по разным атомам кислорода с ионом гидроксония H3O+;
а) и б) – протонизация по алкоксильному и гидроксильному атому кислорода, соответственно
Обе реакции экзотермические, но в первом случае тепловой эффект реакции почти в три раза меньше (ДНа= -120.2 кДж/моль, ДGа = -175.5 кДж/моль) чем во втором (ДНа= -370.9 кДж/моль, ДGа = -420.3 кДж/моль). Скорость превращения протонированного по гидроксиль-ному атому кислорода ГПК должна быть значительно больше, что согласуется с эксперимен-тальными данными.
Аналогичные результаты были получены и в случае протона, сольватированного ацето-нитрилом (рис. 3). Из приведенных данных видно, что протонизация гидропероксида по гидроксильному атому кислорода (О2) энергетически более выгодна, величина ДG значитель-но меньше, чем в случае протонизации алкоксильного атома кислорода (О1). Если принять, что образование целевых продуктов (фенола и ацетона) происходит исключительно через катион II, то полученные результаты согласуются с результатами экспериментов: при кислот-ном катализе основная часть ГПК разлагается на фенол и ацетон.
|
|
a) ДHр = -151.3 кДж/моль, ДGр = -106.9 кДж/моль | б) ДHр = -394.3 кДж/моль, ДGр = -364.1 кДж/моль |
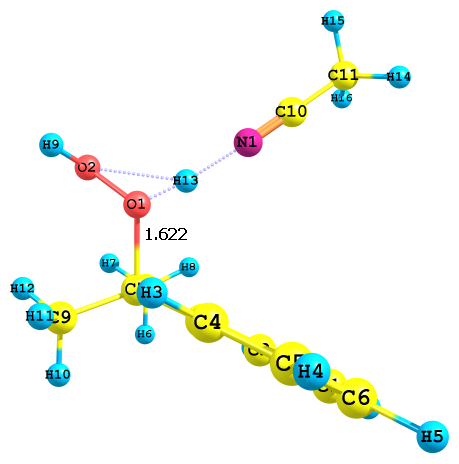
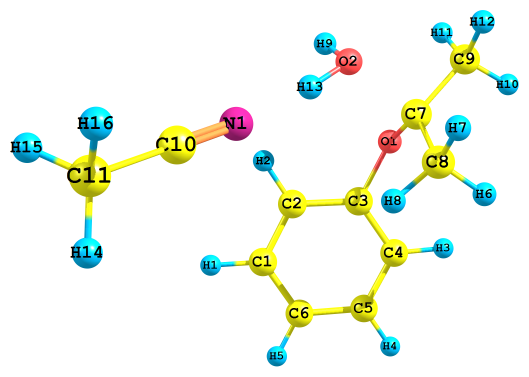
Рис. 3. Результат протонизации ГПК по разным атомам кислорода с сольватированным ацетонитрилом протона; протонизация по а) алкоксильному и б) гидроксильному атому кислорода
|
|
а) ДHр = -134.5 кДж/моль, ДGр = -89.3 кДж/моль | б) ДHр = -121.5 кДж/моль, ДGр = -78.8 кДж/моль |
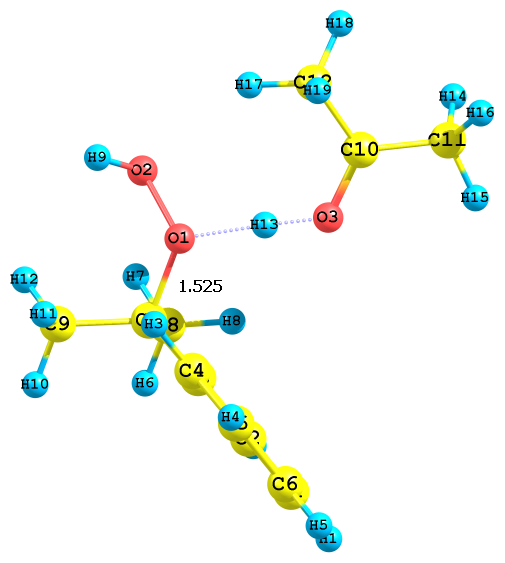
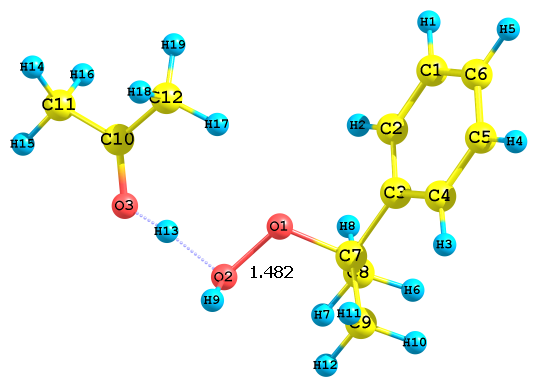
Рис. 4. Результат протонизации ГПК по разным атомам кислорода с сольватированным ацетоном протона; протонизация по а) алкоксильному и б) гидроксильному атому кислорода
При растворении серной кислоты в сильно основных растворителях (ДМСО, формамид, этанол и т. д.) пересольватация протона гидропероксидом затруднена (рис. 5), соответственно образование катионов, ведущих цепь, не происходит. Экспериментально установлено, что в разбавленных растворах серной кислоты (5.10-3 моль/л) при температуре 50 oС в ДМСО, фор-мамиде, этаноле в течение 3 часов заметное разложение ГПК не происходит [14].
|
|
а) ДHр = -117.7 кДж/моль, ДGр = -71.2 кДж/моль | б) ДHр = -114.4 кДж/моль, ДGр = -68.3 кДж/моль |
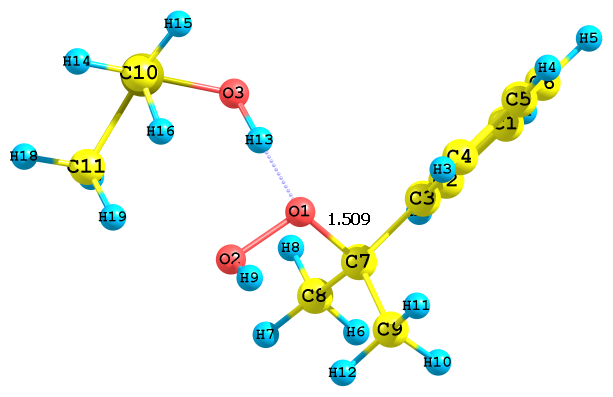
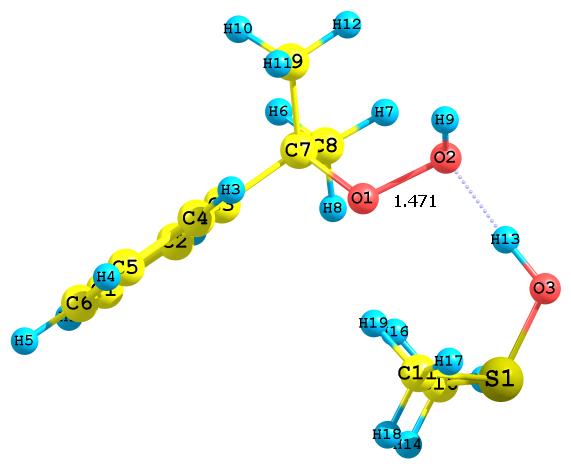
Рис. 5. Предреакционные комплексы, образующиеся при взаимодействии протонированного диметилсульфоксида с а) алкоксильными и б) гидроксильными атомами кислорода ГПК
На качественном уровне значения ДGреакции, а также длины связей Cum-O1 и O1–O2 позволяют оценить скорость реакции с сольватированным различными растворителями протоном. Так, например, для структуры рис. 3б (в ацетонитриле) скорость реакции будет выше, чем в случае структуры на рис. 4б (в ацетоне) 5б (в ДМСО). Протонизация по гидроксильному атому кислороду идет быстрее, чем по алкоксиольному, поскольку длина связи Cum-O1 лишь незначительно увеличивается по сравнению с таковой в ГПК. Сольвати-рованный с различными растворителями протон, видимо, обладает различной активностью по отношению к алкоксильному атому кислорода: наблюдается корреляция между длиной связи Cum-O1 гидропероксида и активностью протона, сольватированного молекулами воды, ацето-нитрила, ацетона и ДМСО. Наибольшая длина связи оказалась в случае иона гидроксония. Поэтому можно предполагать, что энергия активации этого процесса будет наименьшая. В дальнейшем планируется более детально изучить реакции превращения ГПК в присутствии различных протонированных растворителей. Полученные данные соответствует эксперимен-тальным результатам: реакция разложения ГПК в ацетонитриле идет достаточно быстро, а в идентичных условиях в ацетоне медленно, а в ДМСО практически не протекает.
Необходимо отметить, что в разбавленных водных растворах сильных кислот (H2SO4, HCl) заметное разложение ГПК не происходит, что связано образованием слабых кислотных частиц типа Н5О2+:
Схема 4
![]()
При больших концентрациях воды в растворе преобладает ион H5O2+, возможно обра-зуются более сложные комплексы сольватированного протона. Нами на данном этапе рас-смотрены реакции с участием H5O2+. Моделирование взаимодействия этого иона с гидро-пероксидом показало, что при этом протонизация ГПК несколько затруднена, что подтверж-дается отсутствием заметного кислотного разложения ГПК в сильно разбавленных водных растворах H2SO4. Не зависимо от того, какой атом кислорода пероксидной группы подвер-гается атаке катионом H5O2+, величина ДG комплексов очень близки, после протонизации дальнейшего изменения длины связи Cum-O1 и O1–O2 не происходит (рис. 6).
|
|
а) ДHр = -30.6 ккал/моль, ДGр = -19.6 ккал/моль | б) ДHр = -32.1 ккал/моль, ДGр = -21.2 ккал/моль |
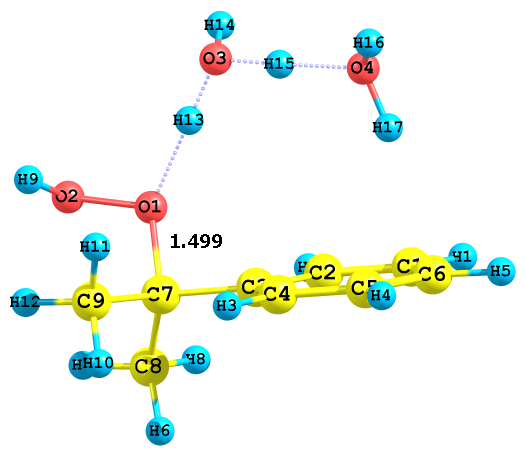
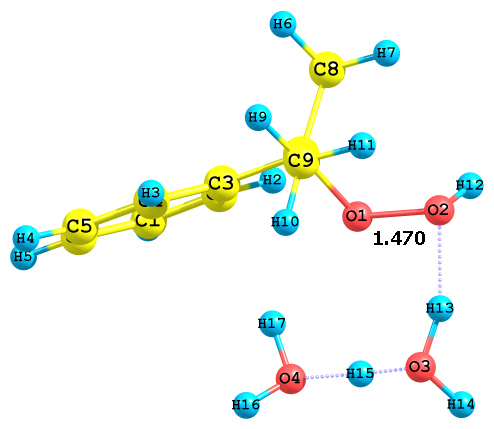
Рис. 6. Предреакционные комплексы, образующиеся при взаимодействии
H5O2+ с алкоксильными и гидроксильными атомами кислорода ГПК
Результаты квантово-химического исследования показывают, что протонированный ГПК по гидроксильному атому кислорода перегруппируется путем отщепления молекулы воды с синхронной миграцией алкоксильного атома кислорода, как показано ниже (рис. 7):
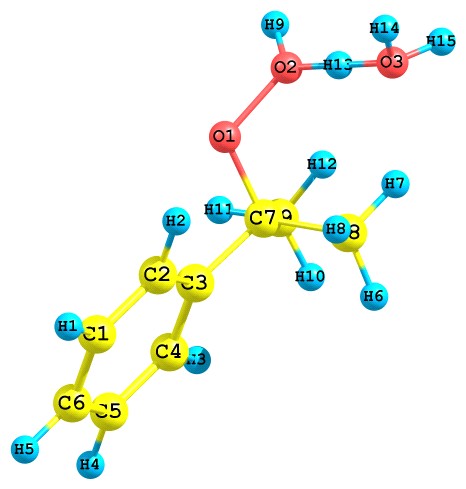
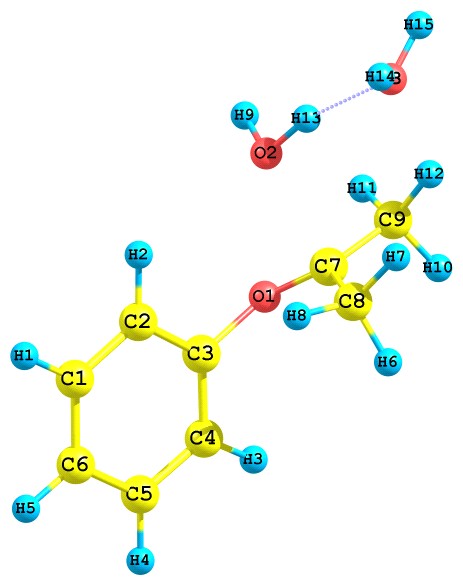
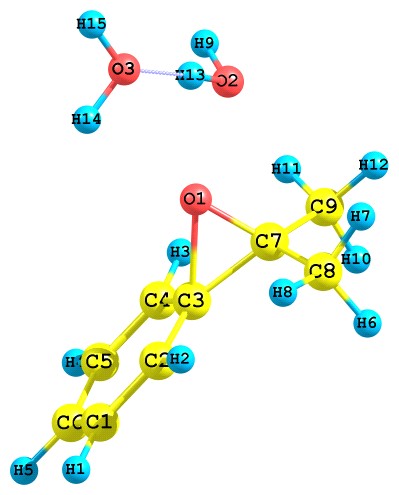
Рис. 7. Превращение протонированного ГПК по гидроксильному атому кислорода
Далее образующийся катион II – взаимодействует со второй молекулой гидропероксида (или водой) с образованием комплекса III (см. схема 2), который далее превращается в фенол и ацетон. В случае протонизации алкоксильного атома кислорода возможно отщепление пер-оксида водорода, с образованием карбокатиона. Результаты этого исследования будут изло-жены в следующих сообщениях.
Заключение
Найдено, что неассоциированная мономерная форма гидропероксида кумола имеет не-сколько конформаций, получаемых при вращении вокруг связей С–О и О–О. Энергетически наиболее выгодной является конфигурация, имеющая молекулярную водородную связь типа Н…π, образующуюся между атомом гидропероксидного водорода и π-электронами бензоль-ного ядра. В образовании фенола и ацетона участвует другая конформация гидропероксида, в которой фрагмент O–O–H развернут в противоположную сторону относительно бензольного кольца и после протонизации этой конфигурации начинается разложение гидропероксида кумола.
Протонизация гидропероксида возможно по обоим атомам кислорода. В случае прото-низации гидроксильного атома кислорода отщепляется вода с синхронным образованием кар-бокатиона, ведущая цепь дальнейшего превращения гидропероксида в сторону фенола и ацетона, а в случае алкоксильного атома кислорода возможно отщепление пероксида водоро-да с образованием карбокатиона, который в итоге превращается в побочные продукты.
Скорость протонизации гидропероксида контролируется параметрами среды, в поляр-ных и высокоосновных растворителях протонизация затруднена.
Полученные результаты и их согласие с экспериментальными результатами дает воз-можность провести дальнейшее исследование квантово-химическими методами основные стадии кислотно-каталитического разложения гидропероксида кумола.
Экспериментальная часть
Расчеты производились программой Gaussian03 гибридным методом теории функционала плотности B3LYP. В качестве предварительного использовался базисный набор 6-31G(d), полученные результаты уточнялись в расширенном базисном наборе 6-311++G(df, p). Расчет производился для газовой фазы, т. е. мы имеем дело с отдельной молекулой, не испытывающей никакого влияния со стороны соседних молекул (современные квантово-химические методы исследования в жидкой или твердой фазе, к сожалению, еще довольно ненадежны). В ходе расчетов проводилась полная оптими-зация всех геометрических параметров молекул. Соответствие найденных структур минимумам энер-гии и переходным состояниям доказывалось соответственно всеми положительными собственными значениями или наличием одного отрицательного собственного значения матрицы Гессе.
В качестве стартовой точки при поиске переходных состояний служили либо результаты известных работ, либо структуры, составленные по «химической интуиции». После локализации переходного состояния во всех случаях производились спуски (IRC) из переходного состояния в сторону продуктов и в сторону реагентов с шагом 0.02–0.03 Е по поверхности потенциальной энергии. После завершения спусков с последней найденной точки производился досчет (оптимизация) для уточнения конформации реагентов (и продуктов реакции), и структуры предреакционного комплекса (в случае бимолекулярных реакций).
Выводы
Моделирование элементарных стадий кислотно-каталитического разложения квантово-химическими методами позволило выявить тонкий механизм протонизации гидропероксида и процессов его дальнейшего превращения в различных растворителях, что может сыграть неоценимую роль в прогнозировании влияния среды и добавок в превращениях гидроперок-сидов и, следовательно, в регулировании скорости и селективности процесса кислотно-ката-литического разложения гидропероксида кумола.
Литература
, Голованенко получение фенола и ацетона. М.: Госхимиздат. 1963. 191с. Kharash M. S., Fono A., Nudenberg W. J. Org. Chem. 1950. No.15. P.748. F. H. Seubold, W. E. Vaughan. J. Am. Chem. Soc. 1953. No.75. P.3790. Механизм кислотного разложения гидроперекисей. Кинетика и катализ. 1960. Т. I. Вып.3. С.365-373. , ДАН СССР. 1959. Т.128. С.341. , , Грагеров образования и разложения гидроперекиси кумола в среде Н2О18. Журнал общей химии. 1962. Т.32. С.758-760. , , Майзус среды в радикально-цепных реакциях окисления органических соединений. М.: Наука. 1973. 279с. Фиальков как средство управления химическим процессом. Л.: Химия. 1990. 240с. ротон в химии. М.: Мир. 1974. , Моисеев некоторых реакций с участием перекисей. Успехи химии. 1960. Т.29. №4. С.425. , Куликов кинетики и механизма разложения гидроперекиси изопропилбензола, катализируемого гетерополикислотами в ацетоне. Кинетика и катализ. 1981. Т.22. Вып.4. С.956-962. Sheldon R. A., Doorn J. A. Observation by PMR Spectroscopy of the Intermediate alkoxycarbonium ions in the Acid-catalized Decompozition of Organic Hydroperoxides. Tetrahedron Letters. 1973. Р.1021. Закошанский процесс кислотно-каталитического разложения гидропероксида кумола. Катализ в промышленности. 2002. №5. С.9-22. Закошанский и кинетика кислотно-катализируемого разложения гидропероксида кумола. Катализ в химической и нефтехимической промышленности. 2004. №4. С.3-15. Закошанский образования димеров α-метилстирола и орто-, паракумилфенолов. Катализ в химической и нефтехимической промышленности. 2005. №1. С.3-11. Мамедов процессом кислотно-каталитического разложения гидропероксида кумила с помощью растворителей. Дисс. канд. техн. наук. Казань: Казанс. гос. технол. ун-т. 1993. 130с. , , Винник разложения гидропероксида кумола в водных растворах HCl. Кинетика и катализ. 1988. Т.29. Вып.6. С.1471-1474. , , Харлампиди Льюиса как катализаторы гомолиза гидроперекисей. Изв. АН СССР. 1975. Т.223. №6. С.1412-1415. , , Жидкова катализ в радикальном распаде гидроперекисей. Изв. АН СССР. Сер. хим. 1976. №7. С.1486-1489.