

Партнерка на США и Канаду по недвижимости, выплаты в крипто
- 30% recurring commission
- Выплаты в USDT
- Вывод каждую неделю
- Комиссия до 5 лет за каждого referral
Публикация доступна для обсуждения в рамках функционирования постоянно
действующей интернет-конференции “Бутлеровские чтения”. http:///readings/
Поступила в редакцию 23 октября 2006 г. УДК 54.02+519.852.6.
Использование SAR и QSAR-методологии для прогнозирования свойств супертоксикантов. Программный комплекс COMSAT
для установления количественных соотношений структура-активность органических соединений, проявляющих
субстратную специфичность к семейству CYP450 человека.
© * и +
Кафедра органической химии и биологической химии. Биолого-химический факультет.
Марийский государственный университет. Пл. Ленина, 1, корпус А. г. Йошкар-Ола, 424001. Россия. Тел.: (8362) 42-32-79. E-mail: *****@***ru
_______________________________________________
*Ведущий направление; +Поддерживающий переписку
Ключевые слова: QSAR, молекулярные дескрипторы, цитохром Р450, диоксиноподобные супертоксиканты.
Аннотация
В настоящей работе предложена методология для решения задачи прогнозирования структура-свойство органических соединений, на основе оценки их субстратной специфичности к энзимам семейства Р450 и путей деградации, а также построения семейств прогностичных QSAR-уравнений с использованием в качестве дескрипторов электронных, физико-химических, топологических и геометрических параметров.
Введение
Анализ взаимосвязи между структурой органических соединений и их биологической активностью в настоящее время является ключевой проблемой в теоретических и токсико-логических исследованиях. Уже более 40 лет QSAR парадигма находит свое практическое применение в фармацевтической химии, агрохимии, токсикологии и скрининге лекарствен-ных средств. Основные положения этой парадигмы базируются на представлении некоторого рода активности как функции структуры от электронных параметров, гидрофобности и стерических факторов. И надо заметить, что результаты QSAR-исследований хотя и сохраняют, в общем, свою оригинальную сущность, но зачастую имеют механистическую интерпретацию. Большинство широко используемых критериев отбора семейств прогностич-ных QSAR-уравнений основываются на оценке их предсказательной способности [1].
Следует заметить, что в настоящее время сформировалось два фундаментальных подхо-да к решению задачи анализа структура-биологическая активность/токсичность исходя из молекулярной структуры химического соединения – первый подход основан на использо-вании экспертных систем, а второй подход реализуется на основе принципа линейности свободных энергий.
Экспертные системы представляют собой коллекцию правил, полученных при исследовании уже существующих объектов. Собственно представляемая химическая струк-тура проходит идентификацию при помощи имеющегося набора правил, а затем выдается решение. При этом если система правил формируется человеком, то говорят о человеческой экспертной системе. Если же экспертная система способна самостоятельно формировать классификационные правила без участия человека, то она является машинно-обучаемой. Недостатком человеческих экспертных систем является то, что они могут быть сформированы исходя из статистически незначимых наблюдений, быть совершенно неподходящими для обучаемых целей, особенно, если в экспертной системе отсутствуют диагностические проце-дуры коррекции ошибок. И, кроме того, представляемые данные могут вносить допол-нительный шум, как следствие внесения неотфильтрованных экспериментальных данных. Однако самый большой недостаток, присущий человеческим экспертным системам, заключа-ется в преимущественном описании только «положительных» правил, допуская при этом, что все химические соединения, не попадающие под эти правила не проявляют значительной активности, что в конечном итоге ведет к большому числу ошибочных классификаций.
Для искусственных экспертных систем характерна объективность при формировании классификационных правил, но они в свою очередь могут отражать неправильные соотношения структура-биологическая активность/токсичность для химического соединения, особенно, когда классификационные правила формируются исходя из наличия или отсутствия определенной атомной группировки. Кроме того, наиболее часто используемые индикаторные функции не обладают возможностью адекватного описания электронной структуры и вместо них рекомендуется использовать значения электротопологического состояния, которые способны описывать различия в электронных свойствах молекулярного фрагмента или атомной группировки [2].
Учитывая недостаточность априорной информации на основании атомных и фрагмент-ных вкладов весьма непросто выработать правила и критерии, для установления соотношений структура-свойство. Классический подход, основанный на сравнительном анализе молекуляр-ных полей (CoMFA) позволяет идентифицировать биологическую активность при исполь-зовании физико-химических дескрипторов или индикаторных переменных. При этом основ-ным условием является сродство молекулярной структуры к участку связывания. Данный подход позволяет сформировать набор правил, направленных на идентификацию субстратной специфичности, и это в свою очередь дает возможность сформировать дерево решений, позволяющее отделять одни субстраты от других, тем самым, определяя характер их биологического действия.
Результаты и их обсуждение
В рамках разрабатываемого нами программного комплекса COMSAT (Computer Optimized Molecular Structure-Activity Testing) для получения количественной характеристики структура-биологическая активность, а также метаболитических профилей органических соединений было предложено использовать комбинированный подход, основанный на сочетании возможностей классического сравнительного анализа молекулярных полей (CoMFA), человеческой экспертной системы и эмпирических линейных соотношений. Блок-схема программного комплекса приведена на рис. 1. Основные характеристики и сравнение с другими известными программными комплексами представлены в табл. 1.
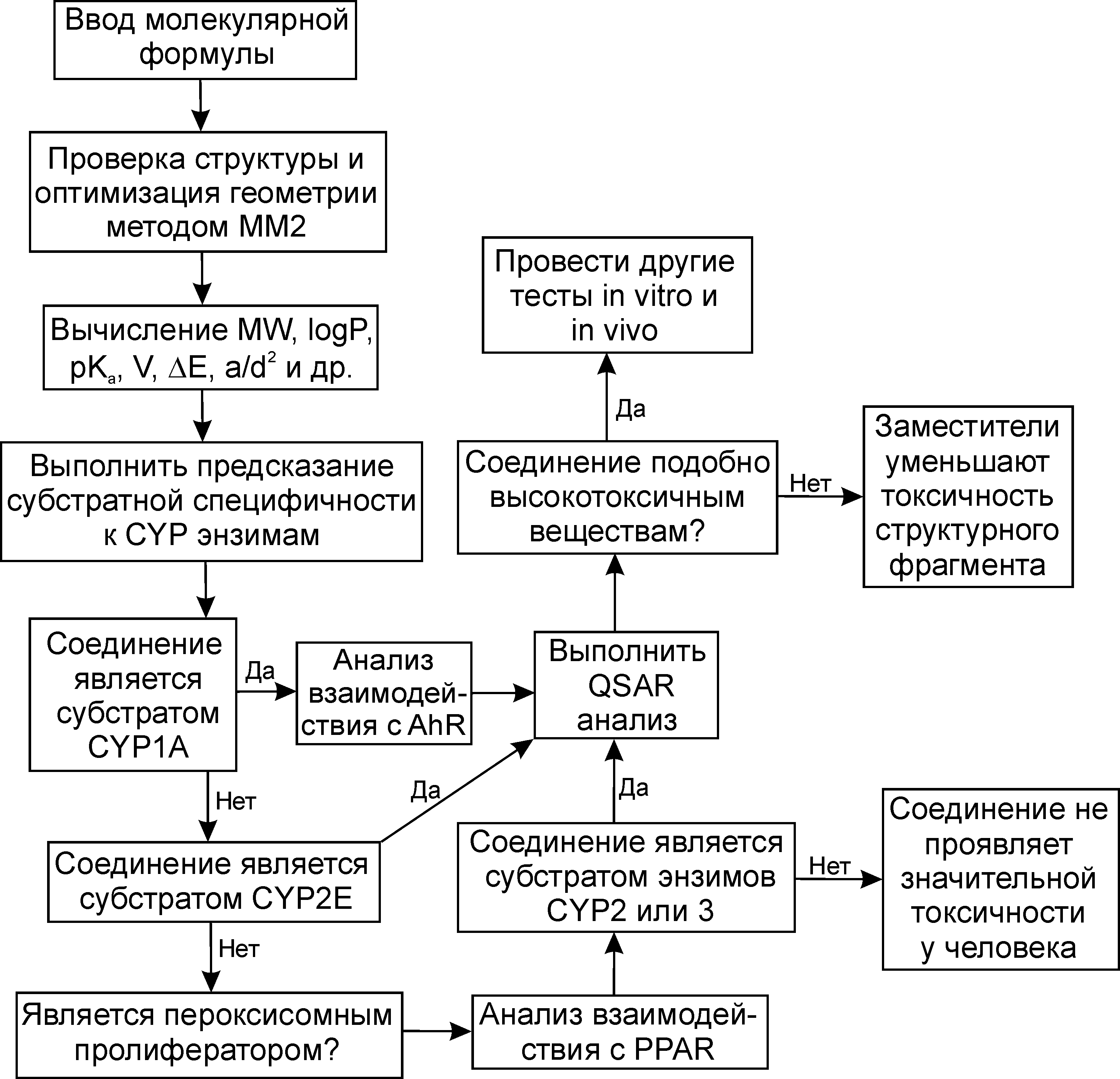
Рис. 1. Блок-схема оценки структура-активность органических соединений
Табл. 1. Сравнение пяти компьютерных программ по оценке токсичности [3]
Название | COMSAT | COMPACT | DEREK | Hazard-Expert | TOPKAT |
Молек. ст-ра | 3D | 3D | Только 2D | Только 2D | Только 2D |
Электр. структура | + | + | - | - | Расчёт эмпирич. зарядов |
Метаболизм | + | + | - | + | - |
Log P | + | - | - | + | + |
pKa | + | - | - | + | + |
Молек. взаимод. | + | + | - | - | - |
QSAR | + | + | - | - | + |
Корреляция с токсичностью | Не известно | ~70% | Не известно | Не известно | 95% |
ЭВМ | PC | Любая | VAX/ Unix | PC | PC |
Стоимость | Не известно | 1000$ за соед. | Не известно | 15000$ | 15000$/ модуль |
Число атомов | Огранич. PC | 350 | 64 | Огранич. PC | Огранич. PC |
Основной алгоритм оценки | QSAR/ Взаимод. с Р450 | Взаимод. с Р450 | FDA | EPA | QSAR/ NCI |
Публикации | 5 | >30 | >5 | >5 | >10 |
Лет в использ. | Нет | 10 | 7 | 7 | 10 |
Коммерческая | Нет | Нет | Да | Да | Да |
Сокращения: 3D, трёхмерная структура; 2D, двумерная структура; +, имеется метод вычисления;
-, нет метода вычисления; QSAR, количественная оценка структура-активность; FDA/NCI,
Food and Drug Administration/National Cancer Institute; PC, персональный компьютер.
1. Молекулярная структура
В разрабатываемом нами программном комплексе COMSAT, используется современный графический интерфейс для ввода информации о химическом соединении в виде трехмерной модели, ставший de facto стандартом химических программ и позволяет пользователю вводить целевую структуру в произвольной последовательности. При этом химическая структура в данном программном комплексе представляется в виде таблицы связности, состоящей из таблицы атомов и таблицы связей, а затем подвергается канонизации. Минимизация энергии трехмерной структуры химического соединения производится в картезианских координатах при помощи молекулярной механики – методом Ньютона. Практика показывает, что молекулярная механика дает более реалистичный результат, чем при использовании ряда популярных полуэмпирических методов. Это отчасти объясняется тем, что для параметризации используются экспериментальные данные, полученные на реальных моделях, однако такой подход оказывается неоправданным в случае наличия весьма экзотических молекулярных систем. В качестве параметров силового поля могут использоваться ММ3 (2000) с коррекцией на электроотрицательность и водородную связь, а также ММ2 (1991), тоже включающий π-системы, но в нем отсутствует поправка на аномерный эффект и коррекция электроотрицательности. Данная часть программного комплекса, отвечающая за оптимизацию геометрии молекулярной структуры, была реализована нами в виде отдельного dll-модуля, созданного на основе переработанных исходных текстов свободно распространяемого пакета Tinker [4].
Кроме того, возможен обмен информацией через файл с рядом известных квантово-химических программ. Конвертирование из одного формата файла в другой было также реализовано в виде dll-модуля, созданного на основе переработанных исходных текстов свободно распространяемого программного комплекса Babel [5].
2. Молекулярные дескрипторы
Установление зависимости структура-свойство основано на построении семейства прогностичных QSAR-уравнений и основывается на теории обучаемых систем. При этом общем случае, задача обучения по прецедентам сводится к восстановлению функциональной зависимости заданной выборки структура-свойство, т. е. на установлении взаимосвязи между биологической активностью и параметрами химического соединения, представленных в виде независимых переменных, которые в свою очередь получили название молекулярных дескрипторов. В QSAR могут быть использовано более 3000 молекулярных дескрипторов. При этом принято подразделять на 2D (двумерные) и 3D (трехмерные) молекулярные дескрипторы. К 2D дескрипторам относятся те, которые не используют информацию о трехмерных характеристиках химического соединения. Существует несколько способов классификации дескрипторов, но наиболее часто употребляемой классификацией является учет природы эффекта. Например, электронные, стерические, межмолекулярные эффекты соответствуют аналогичным дескрипторам – электронным, топологическим, физико-хими-ческим и т. д. Нужно заметить, что не существует общего правила для подбора дескрипторов, а выбор того или иного дескриптора зависит от специфики решаемой задачи. В целом дескрипторы должны иметь ясный физический смысл, обладать предсказательной способ-ностью и обеспечивать возможность идентификации связей [6]. Используемые нами моле-кулярные дескрипторы используются не только для построения семейств QSAR-уравнений, но и для оценки субстратной специфичности по отношению к энзимам семейства Р450. Из дескрипторов элементного уровня нами используется значение молекулярного веса MW, получаемого непосредственно из списка атомов (брутто-формулы) молекулярной структуры.
На основе матрицы расстояний и Ван-дер-Ваальсового пространства нами используется ряд молекулярных дескрипторов. Геометрический индекс Винера GIW рассчитывался как полусумма недиагональных элементов матрицы расстояний для оптимизированной геометрии химической структуры. Индекс Платта F находится суммированием для каждой вершины количества их смежных вершин. Значение индекса Балабана рассчитывали на основе соотношений описанных в [7, 8]. Ван-дер-Ваальсовую поверхность, площадь молекулы и молекулярный объем находили при помощи реализованных алгоритмов для оптимизирован-ной молекулярной структуры [9, 10]. Максимальную длину молекулы (Lmax) определяли также для оптимизиованной геометрии как прямое расстояние между крайними атомами молекулы.
Коэффициент распределения октанол-вода (logP) является важным параметром, опреде-ляющим биоперенос вещества и его доступность к деградации микросомами печени. Значения logP рассчитывали методом Реккера на основании фрагментных и атомных вкладов [11]. Значения pKa рассчитывались исходя из уравнений Тафта и Гаммета на основании парамет-ров заместителей, а также уравнения Дьюара-Грисдаля для полициклических ароматических соединений. Фрагментные и атомные вклады, а также параметры заместителей были взяты из литературных данных.
Квантово-химические дескрипторы (энергии граничных орбиталей, поляризуемость и др.) рассчитывали при помощи пакета MOPAC 6.0 методом РМ3 для молекулярных систем с оптимизированной геометрией методами молекулярной механики [12].
3. Предсказание субстратной специфичности
Учитывая тот факт, что органические соединения практически всех классов подвер-гаются ферментативным аэробным превращениям и у млекопитающих около 90% общего метаболизма фазы I происходит с участием цитохромов семейства Р450, становится очевид-ной важность учета субстратной специфичности при изучении токсикологических профилей органических соединений.
Нами была исследована выборка из 244 органических соединений различных классов. Субстраты подсемейства CYP1А1 были представлены 81 соединением, CYP1А2 – 12 соеди-нений. Семейство CYP2А представлено выборкой из 12 соединений в качестве субстратов, CYP2В – 26, CYP2С – 17, CYP2D – 32, CYP2Е – 30 и CYP3А – 34 представителей. Для всех исследуемых соединений была проведена оптимизация геометрии методом Ньютона с параметрами силового поля ММ2. Учитывая, что физиологические лиганды индуцируют отличные от CYP1, CYP2, CYP3 изоформы P450, то они должны быть идентифицированы в первую очередь. Для этого достаточно сформировать базу данных наиболее важных экзогенных субстратов иных изоформ CYP, чем CYP1, CYP2, CYP3 и производить структурный поиск. Субстратов 1А1 от остальных CYP отличает планарная геометрия, ароматический характер и разность ВЗМО-НСМО в пределах 7.8-8.5 eV. Субстратами 1А2 являются планарные ароматические азотсодержащие соединения, где азот является заместителем или входит в состав циклa. Индукция обеих изоформ CYP контролируется Ah-рецептором. Значения молекулярного объема (Е3) субстратов 2Е лишь немного не дотягивают до интервала 95%: 44.20 < 69.72 < 72.48 и могут быть отделены от субстратов 3А: 234.02 < 251.61 < 269.20, где наблюдается существенный разброс значений объема. Наибольшие трудности возникают при разделении субстратов 3А и 2A, B,C, D, где для последних имеется лишь тенденция группироваться вокруг среднего 186.71 Е3 с доверительным интервалом существенно меньшим 95%. Значения pKa субстратов CYP2С с доверительным интервалом 2.22 < 4.83 < 5.27, а CYP2D: 9.34 < 9.78 < 10.22 не обнаруживают статистической значимости на уровне 95%. Субстраты 2А и 2В как правило не имеют кислотных или основных групп и являются нейтральными. Значения logP субстратов CYP2B не укладываются в довери-тельный интервал 95%: 3.65 < 4.31 < 4.96. Аналогичная ситуация наблюдается и для субстратов CYP2А: -0.11 < 0.36 < 0.72. Область пересечения наблюдается для 2А: 1.46, а для 2В: 1.41, где надежная идентификация субстратов невозможна.
Таким образом, на исследованных выборках соединений обнаруживается приблизитель-ная оценка субстратной специфичности. Но, несмотря на трудность разнесения субстратов 3А и 2A, B,C, D – данные интервальные оценки могут использоваться для классификации субстра-тов (рис. 2). Надо отметить, что некоторые эндогенные лиганды могут являться субстратами нескольких представителей семейства Р450. Кроме того, наблюдаются существенные разли-чия между отдельными представителями монооксигеназных систем человека и других мле-копитающих. Данные различия не позволяют в полной мере переносить данные по токсичности от экспериментальных животных к человеку.
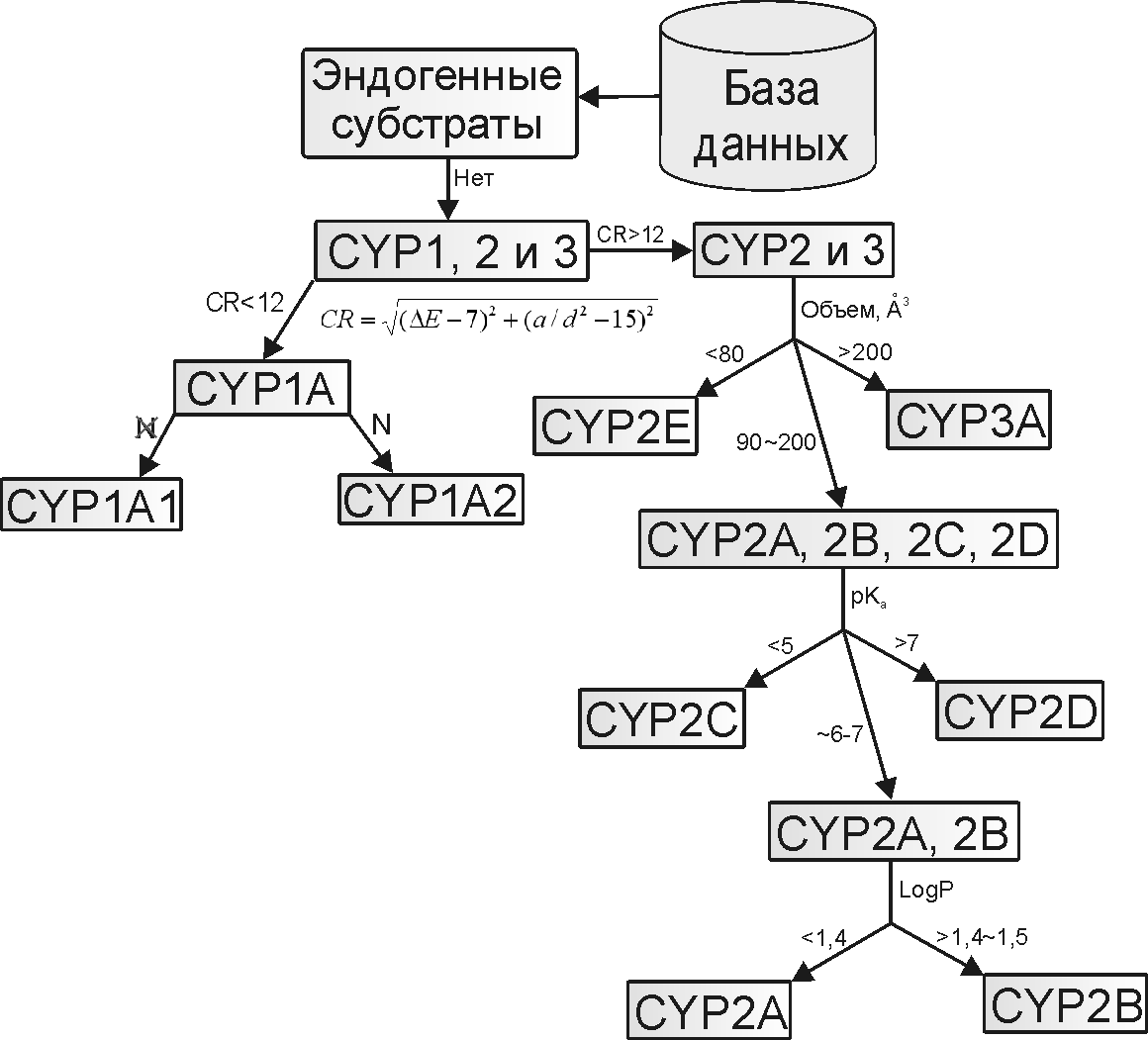
Рис. 2. Схема идентификации субстратов CYP450 человека
4. Оценка метаболитического профиля
Кроме того, нами сформирована система эмпирических правил, позволяющая предска-зать пути превращения субстратов монооксигеназной системы микросом млекопитающих. Монооксигеназные реакции, катализируемые семейством энзимов Р450 млекопитающих включают в себя гидроксилирование алифатических соединений, деалкилирование, окисление у гетероатома, образование ненасыщенных соединений, эпоксидирование, окислительную миграцию функциональных групп, а также другие виды превращений. Данные превращения основаны на взаимодействии субстрата с очень реакционноспособной частицей Fe+2(O2) Substrate, входящей в состав порфирина, которая способна отщеплять атом водорода или π-электрон, а также осуществлять рекомбинацию радикалов [13].
Ниже приведены пять основных правил превращений субстратов фазы I на монооксиге-назных системах и, кроме того, имеется возможность формирования дополнительных правил, специфичных для отдельных классов органических соединений.
Правило 1: гидроксилирование у атома углерода:

Правило 2: отщепление гетероатома:
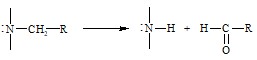
Правило 3: окисление гетероатома:
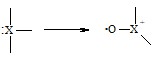
Правило 4: эпоксидирование:

Правило 5: миграция групп непредельных соединений:
На стадии детоксикации фазы II промежуточные электрофильные метаболиты превра-щаются в водорастворимые нетоксические продукты при помощи энзимов семейства глутатионтрансферазы (GSTM), УДФ-глюкуронсульфотрансфераз (UDF), N-ацетилтрансфераз (NAT).
5. Построение прогностичных QSAR-зависимостей
Установление количественной связи структура-активность органических соединений основано на использовании теории обучаемых систем, где качество применяемых алгоритмов, синтезированных по конечным выборкам, является жизненно важной проблемой. В общем случае задача обучения по прецедентам сводится к восстановлению функциональной зависи-мости заданной выборки структура-свойство. Таким образом, нужно построить алгоритм, который бы выдавал адекватные ответы на предъявляемые объекты. Восстановление зависи-мостей по выборкам ограниченного объема реализуется по одной математической схеме – минимизации среднего риска [14].
Для получения хорошей QSAR модели, содержащей как можно меньше независимых переменных, необходимо использовать наиболее подходящие для данного типа объектов алгоритмы классификации. Выбор алгоритма классификации является ключевым моментом в оценке количественных соотношений структура-биологическая активность. Под классифика-цией понимается процесс разбиения исходного набора данных на соответствующие группы. При этом алгоритм классификации должен обладать возможностью предложить модель, способную давать адекватное описание исследуемого объекта. Наиболее используемые алгоритмы классификации, используемые в QSAR можно найти в литературе [15]. Включение минимального числа переменных является необходимым условием для увеличения предсказательной способности математической модели. В противном случае при добавлении переменной не вносится значимой информации, однако, приводя при этом к возрастанию шума и сложности анализа.
Для постройки линейно зависимых параметров нами были реализована МНК-оценка используемая во множественном регресионном анализе и в комбинаторном методе группо-вого учета аргументов (МГУА) [16, 17].
Нами были использованы следующие методы поиска наилучшего подмножества предикторных переменных: метод исчерпывающего поиска подмножества переменных, шаго-вый метод Эйфромсона, метод последовательной замены, метод двухкратной последователь-ной замены, метод прямого включения переменных, метод прямого исключения переменных а также метод поиска наилучшего подмножества на основе критерия Малоуза с коррекцией Джилмура. Кроме того, в качестве критерия на включение/удаление предикторной перемен-ной может использоваться F-критерий, критерий Малоуза, а также современные информа-ционные критерии – критерий Ганнана-Куина и Критерий Шварца. Оценка прогнозирующей способности функции, полученной в результате восстановления зависимости по эксперимен-тальным данным оценивается при помощи процедуры скользящего контроля (удаление 10% тестовой выборки) или при помощи бутстреп-оценки (удаление случайного количества тестовой выборки).
Использование моделей регрессионного типа для прогнозирования свойств, по сравне-нию с часто применяемыми индуктивными методами восстановления функции – методами группового учета аргументов (МГУА) и предельных упрощений (МПУ) имеет то преиму-щество, что индуктивные методы, основываясь на частных фактах не всегда достаточно оптимально характеризуют описываемое свойство. Так что качество индуктивных решений должно определяться не только объяснением отдельных фактов, сколько от экстраполя-ционной способностей этих решений, их возможностью к экспансии в область явления, не охваченную экспериментальными данными. Кроме того, удовлетворительные значения Кри-терия отбора не могут быть достигнуты при малой размерности экспериментальных данных, как следствие невозможности правильного обучения на ограниченной обучающей выборке.
При использовании всех запрограммированных нами методов поиска наилучшего под-множества предикторных переменных для линейного соотношения структура-свойство классов полигалогенированных дибензо-п-диоксинов, дибензофуранов, бифенилов, а также объединенных выборок, кроме метода поиска наилучшего подмножества на основе критерия Малоуза с коррекцией Джилмура получены следующие результаты:
Полигалогенированные дибензо-п-диоксины:
log(1/EC50) = 1 – 446.41176∙HOMO + 3368.58890∙log P
n = 25; r2 : 0.89183; s2ост. = 0.49598; q2 > 0.88;
Полигалогенированные дибензофураны:
log(1/EC50) = 1 + 1870.41498∙log P – 255.67157∙SN + 366.65331∙Lmax
n = 39; r2 : 0.82708; s2ост. = 0.57796; q2 > 0.71;
Полигалогенированные бифенилы:
log(1/EC50) = 1 + 61.40601∙log P + 7465.23809 ∙GIW – 384.80687∙MW
n = 14; r2 : 0.93725; s2ост. = 0.24945; q2 > 0.87.
Результаты QSAR-анализа ряда дибензо-п-диоксинов обнаруживают определяющий вклад параметра log Pow – ответственного за биоперенос токсиканта, а также электронных параметров – HOMO энергии высшей занятой молекулярной орбитали, что в целом отвечает экспериментальным данным. Отрицательное значение последней, согласно теореме Кумпан-са, отвечает значению потенциала ионизации, определяющего донорные свойства экотокси-кантов, что является фактором, контролирующим стабильность соединения в субстрат-рецепторном комплексе.
Факторами, определяющими токсичность полихлорированых дибензофуранов, являются липофильность (log Pow), а также поляризуемость молекулы (SN) – способность деформации электронных оболочек, что является важным параметром при стэкинге.
Для высокохлорированных бифенилов определяющим фактором соотношения струк-тура-активность является липофильность (log Pow) и разветвленность молекулярной струк-туры. При этом следует учесть, что планарная геометрия бифенилов, необходимая для взаимодействия с рецептором, не является энергетически выгодной, существует в различных конформациях, что дает сильное ограничение для использования такого рода моделей.
Попытка построения объединенной модели для полигалогенированных дибензо-п-диоксинов и дибезофуранов не дает прогностичных зависимостей структура-свойство и имеет линейный тренд на графике зависимости остатков от подобранных значений. То же самое справедливо и для объединенной модели, включающей все три класса соединений.
Заключение
Результаты моделирования структура-биологическая активность в рамках предложен-ного программного комплекса показывают определяющую роль молекулярных дескрипторов, которые отражают экотоксичность исследованных полигалогенированных экотоксикантов. В этом численном эксперименте показана ключевая роль липофильных характеристик, электро-нодонорных свойств и поляризуемости молекул.
Использование методов сравнительного анализа молекулярных полей (CoMFA) является продуктивным для предсказания субстратной специфичности к цитохромам семейства Р450. Использование данной модели ограничено лишь одним видом млекопитающих Homo sapiens.
Литература
Burger’s Medicinal Chemistry and Drug Discovery. Sixth Edition. Volume 1: Drug Discovery Editedby Donald J. Abraham. John Wiley&Sons, Inc. 2003.
