

Партнерка на США и Канаду по недвижимости, выплаты в крипто
- 30% recurring commission
- Выплаты в USDT
- Вывод каждую неделю
- Комиссия до 5 лет за каждого referral
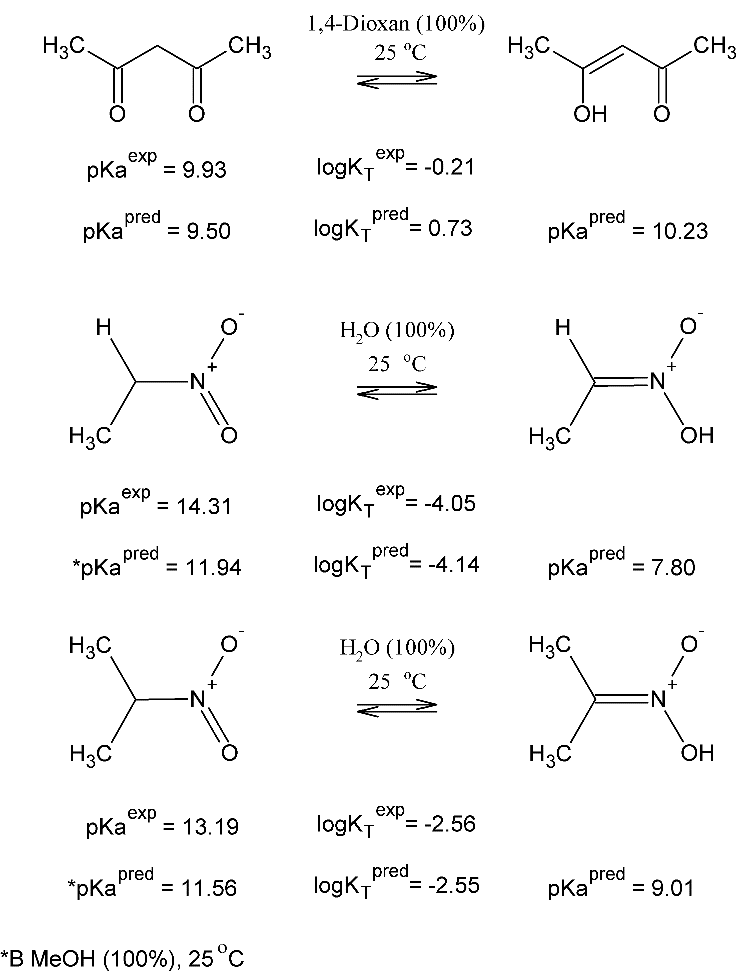
Рисунок 5. Экспериментальные и предсказанные значения logKT и рКа таутомеров компромиссной модели для случаев кето-енольной и нитро-аци таутомерии
Предсказательная способность моделей была также оценена на внешней тестовой выборке, состоящей из 22 реакций (табл. 2). Однако большая часть из них не входили в область применимости модели, в которую вошли только 8 реакций. По данным Табл.2 можно отметить, что качество предсказания для реакций, даже включая те, что находятся вне домена применимости, неплохое, хоть и несколько хуже опубликованной ранее модели c использованием метода SVM [22]. Видимое ухудшение качества совместных моделей по сравнению с индивидуальной обусловлено бoльшим разбросом для точек, находящихся вне домена применимости, Рис. 6. Является ли это недостатком метода обучения или случайностью выяснить в настоящий момент не представляется возможным. На реакциях, находящихся в области применимости модели, ошибка предсказания константы таутомерного равновесия совместной и компромиссной моделью достаточно низкая и близка к таковой для индивидуальных моделей, в том числе и опубликованных нами ранее с использованием нелинейных методов моделирования (SVM и GTM) [23]. На Рис. 6 можно отметить что все модели предсказывают эти точки практически одинаково.
Таблица 2. Значения R2 и RMSE моделей полученные на внешней тестовой выборке
Модель | Все равновесия | Равновесия в области применимости модели | ||
RMSE | R2 | RMSE | R2 | |
Индивидуальная (logKT) | 2.00 | 0.67 | 0.84 | 0.92 |
Лучшая совместная для logKT | 2.37 | 0.53 | 0.83 | 0.92 |
Компромиссная (предсказание logKT и pKa одновременно) | 2.70 | 0.40 | 0.83 | 0.92 |
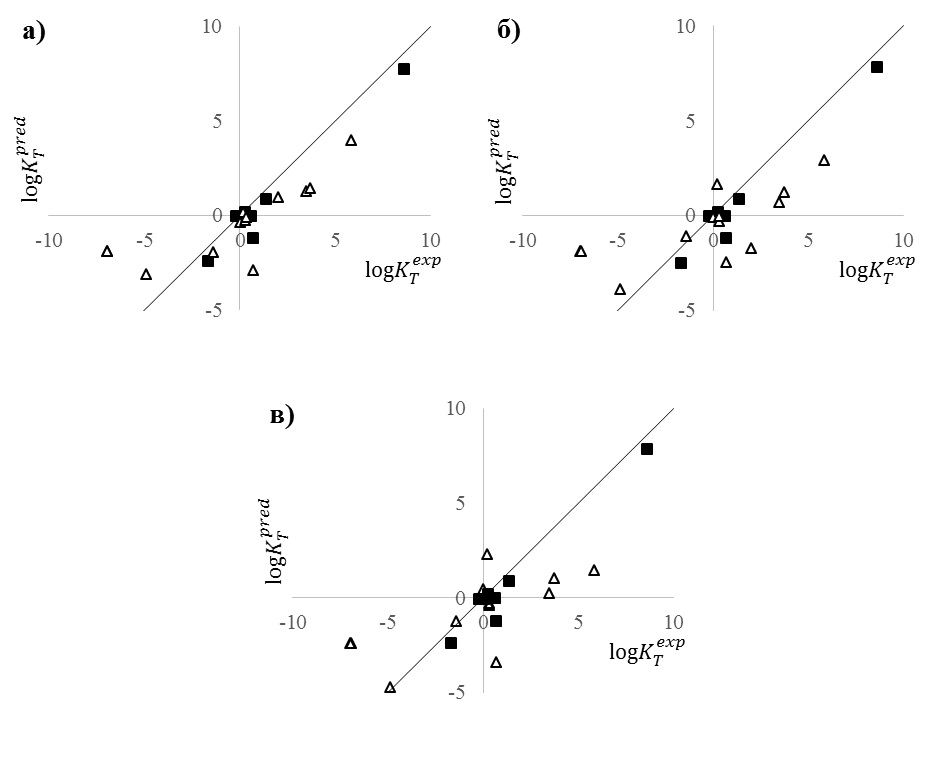
Рисунок 6. Предсказанные и экспериментальные значения logKT для внешней тестовой выборки. Черные квадраты и белые треугольники обозначают предсказания, находящиеся внутри и вне области применимости модели, соответственно. Предсказания получены:
(а) — индивидуальной моделью для logKT, (б) — лучшей совместной моделью для предсказания logKT, (в) — компромиссной моделью.
Выводы
В рамках данной работы нами предложен подход к совместному моделированию и предсказанию двух линейно связанных величин на примере константы кислотности и константы прототропного таутомерного равновесия, см. уравнение (1). Показано, что с использованием метода множественной линейной регрессии с L2 регуляризацией (гребневой регрессии) возможно получение аналитического выражения для расчета регрессионных коэффициентов – выражения (7). При построении данной модели используются одновременно данные по константам равновесия реакций таутомерии и данные по кислотности органических молекул. При этом регрессионные коэффициенты подбираются таким образом, чтобы, с одной стороны, максимально точно воспроизводился показатель кислотности соединений, а с другой, чтобы разность кислотности таутомеров равнялась константе таутомерного равновесия, как того требует уравнение (1).
Было показано, что обучение модели только на данных по кислотности соединений с последующим применением уравнения (1) не может быть использовано для предсказания константы таутомерного равновесия – полученная модель намного хуже нулевой по данным скользящего контроля. Использование же предложенного подхода совместного обучения позволяет даже достигнуть некоторого увеличения в точности предсказания logKT по сравнению с моделью, которая для обучения использует только данные по реакциям таутомерии. При этом полученная модель способна предсказывать кислотности всех таутомеров и разность кислотности будет подчиняться уравнению (1), что требуется термодинамическим циклом, приведенным на Рис. 1. С использованием имеющихся данных по экспериментальному измерению констант кислотности отдельных таутомеров было показано, что предсказанные значения pKa таутомеров согласуются с экспериментальными данными. Качество предсказаний подтверждается с использованием внешней тестовой выборки. При учете области применимости моделей с помощью контроля фрагментов модели совместного обучения и индивидуальные модели демонстрируют одинаковую точность предсказания константы таутомерного равновесия.
Предложенный подход обладает несколькими преимуществами по сравнению с ранее опубликованными нами работами [20–23]. Он способен одновременно предсказывать кислотность соединений, в том числе различных таутомерных форм, и константу таутомерного равновесия практически без потери качества. Он основан на теоретически обоснованном уравнении (1) и поэтому разность кислотности таутомеров равна константе таутомерного равновесия. С его использованием можно оценить процентное содержание таутомерных форм для сложных равновесий с участием молекул, имеющих множество (более 2) таутомеров. С использованием достаточно простых подходов, следующих из термодинамических циклов превращений таутомеров и их кислотности, можно оценивать влияние рН среды на процентное содержание форм без необходимости перестройки модели.
Как существенное ограничение разработанного нами подхода, можно отметить то, что зависимость кислотности от дескрипторов условий (растворителя и температуры) полагается линейной. Вследствие этого построенная модель не описывает в явном виде влияние среды (температуры, растворителя), и поэтому экспериментальные данные, используемые для построения модели, должны быть получены в одинаковых условиях. Для моделирования влияния среды требуется использование нелинейных методов, что является объектом дальнейших исследований.
Благодарности
Исследования выполнены при поддержке Российского научного фонда (соглашение №14-43-00024). Авторы благодарят компанию ChemAxon за предоставление академической лицензии на программное обеспечение JChem.
Литература
[1] M. Sitzmann, W.-D. Ihlenfeldt, M. C. Nicklaus. Tautomerism in large databases. put. Aided. Mol. Des. 2010. Vol. 24., No. 6–7. P. 521–551.
[2] P. Pospisil, P. Ballmer, L. Scapozza, G. Folkers. Tautomerism in computer-aided drug design. J Recept Signal Transduct Res. 2004/02/03. 2003. Vol. 23., No. 4. P. 361–371.
[3] T. Clark. Tautomers and reference 3D-structures: the orphans of in silico drug design. put. Aided. Mol. Des. Springer Netherlands, 2010. Vol. 24., No. 6–7. P. 605–611.
[4] W. Warr. Tautomerism in chemical information management systems. put. Aided. Mol. Des. Springer Netherlands, 2010. Vol. 24., No. 6–7. P. 497–520.
[5] R. A. Sayle. So you think you understand tautomerism? put. Aided. Mol. Des. 2010. Vol. 24., No. 6–7. P. 485–496.
[6] A. R. Katritzky, C. D. Hall, B. E.-D. M. El-Gendy, B. Draghici. Tautomerism in drug discovery. put. Aided. Mol. Des. Springer Netherlands, 2010. Vol. 24., No. 6–7. P. 475–484.
[7] Advanced Chemistry Development Inc. ACD/Tautomers. :version 15.01. Advanced Chemistry Development, Inc., Toronto, ON, Canada. www. , 2015.
[8] ChemAxon. JChem Calculator Plugins 15.8.3. :15.8.3.
[9] Molecular Networks GmbH Computerchemie. MN Tautomer. . Molecular Networks GmbH, Germany and Altamira, LLC, USA. https://www. .
[10] Schrodinger LLC. LigPrep tautomeriser. . Schrodinger, LLC. https://www. /ligprep.
[11] Xemistry GmbH. CACTVS. . Xemistry GmbH. http://www. .
[12] OpenEye Scientific Software. QUACPAC. . OpenEye Scientific Software. https://www. /quacpac.
[13] BIOVIA. BIOVIA Pipeline Pilot. . BIOVIA, USA. http:///products/collaborative-science/biovia-pipeline-pilot.
[14] M. Haranczyk, M. Gutowski. Quantum Mechanical Energy-Based Screening of Combinatorially Generated Library of Tautomers. TauTGen: A Tautomer Generator Program. J. Chem. Inf. Model. 2007. Vol. 47., No. 2. P. 686–694.
[15] N. T. Kochev, V. H. Paskaleva, N. Jeliazkova. Ambit-Tautomer: An Open Source Tool for Tautomer Generation. Mol. Inform. 2013. Vol. 32., No. 5–6. P. 481–504.
[16] J. R. Greenwood, D. Calkins, A. llivan, J. C. Shelley. Towards the comprehensive, rapid, and accurate prediction of the favorable tautomeric states of drug-like molecules in aqueous solution. put. Aided. Mol. Des. Springer Netherlands, 2010. Vol. 24., No. 6–7. P. 591–604.
[17] P. Pospisil, P. Ballmer, L. Scapozza, G. Folkers. Tautomerism in Computer?Aided Drug Design. J. Recept. Signal Transduct. 2003. Vol. 23., No. 4. P. 361–371.
[18] F. Oellien et al. The Impact of Tautomer Forms on Pharmacophore-Based Virtual Screening †. J. Chem. Inf. Model. 2006. Vol. 46., No. 6. P. 2342–2354.
[19] S. J. Angyl, C. L. Angyal. 268. The tautomerism of N-hetero-aromatic amines. Part I. J. Chem. Soc. 1952. P. 1461.
[20] J. Szegezdi, F. Csizmadia. Tautomer generation. pKa based dominance conditions for generating dominant tautomers. The 234th ACS National Meeting, Boston, MA, August 19-23, 2007. Boston, 2007.
[21] F. Milletti et al. Tautomer enumeration and stability prediction for virtual screening on large chemical databases. J. Chem. Inf. Model. 2009. Vol. 49., No. 1. P. 68–75.
[22] T. R. Gimadiev et al. Assessment of tautomer distribution using the condensed reaction graph approach. put. Aided. Mol. Des. 2018. Vol. 32., No. 3. P. 401–414.
[23] M. Glavatskikh et al. Visualization and Analysis of Complex Reaction Data: the Case of Tautomeric Equilibria. Mol. Inform. Wiley-Blackwell, 2018. Vol. 0., No. 0.
[24] , , . Введение в хемоинформатику: учеб. пособие. Ч. 4. Методы машинного обучения. . Казань: Изд-во Казан. ун-та, 2016. 330 p.
[25] A. Varnek, D. Fourches, F. Hoonakker, V. P. Solov’bstructural fragments: an universal language to encode reactions, molecular and supramolecular structures. put. Aided. Mol. Des. Springer Netherlands, 2005. Vol. 19., No. 9–10. P. 693–703.
[26] ChemAxon Standardizer. :15.8.3.0. 2015.
[27] T. I. Madzhidov et al. Consensus approach to atom-to-atom mapping in chemical reactions. Butlerov Commun. 2015. Vol. 44., No. 12. P. 170–176.
[28] Indigo. . https:///ggasoftware/indigo, 2015.
[29] F. Ruggiu, G. Marcou, A. Varnek, D. Horvath. ISIDA Property-labelled fragment descriptors. Mol. Inform. 2010. Vol. 29., No. 12. P. 855–868.
[30] R. Fiorella et al. Individual Hydrogen?Bond Strength QSPR Modelling with ISIDA Local Descriptors: a Step Towards Polyfunctional Molecules. Mol. Inform. Wiley-Blackwell, 2014. Vol. 33., No. 6?7. P. 477–487.
[31] J. Catalan et al. Progress towards a generalized solvent polarity scale: The solvatochromism of 2-(dimethylamino)-7-nitrofluorene and its homomorph 2-fluoro-7-nitrofluorene. Liebigs Ann. WILEY-VCH Verlag, 1995. Vol. 1995., No. 2. P. 241–252.
[32] J. Catalan, C. Diaz. A Generalized Solvent Acidity Scale: The Solvatochromism of o-tert-Butylstilbazolium Betaine Dye and Its Homomorph o, o?-Di-tert-butylstilbazolium Betaine Dye. Liebigs Ann. WILEY-VCH Verlag, 1997. Vol. 1997., No. 9. P. 1941–1949.
[33] J. Catalan et al. A Generalized Solvent Basicity Scale: The Solvatochromism of 5-Nitroindoline and Its Homomorph 1-Methyl-5-nitroindoline. Liebigs Ann. WILEY-VCH Verlag, 1996. Vol. 1996., No. 11. P. 1785–1794.
[34] T. Yokoyama, R. W. Taft, M. J. Kamlet. The solvatochromic comparison method. 3. Hydrogen bonding by some 2-nitroaniline derivatives. J. Am. Chem. Soc. 1976. Vol. 98., No. 11. P. 3233–3237.
[35] M. J. Kamlet, R. W. Taft. The solvatochromic comparison method. I. The. beta.-scale of solvent hydrogen-bond acceptor (HBA) basicities. J. Am. Chem. Soc. American Chemical Society, 1976. Vol. 98., No. 2. P. 377–383.
[36] M. J. Kamlet, J. L. Abboud, R. W. Taft. The solvatochromic comparison method. 6. The. pi.* scale of solvent polarities. J. Am. Chem. Soc. American Chemical Society, 1977. Vol. 99., No. 18. P. 6027–6038.
[37] P. Polishchuk et al. Structure–reactivity modeling using mixture-based representation of chemical reactions. put. Aided. Mol. Des. 2017. Vol. 31., No. 9. P. 829–839.

|
Из за большого объема этот материал размещен на нескольких страницах:
1 2 3 4 |
