
Партнерка на США и Канаду по недвижимости, выплаты в крипто
- 30% recurring commission
- Выплаты в USDT
- Вывод каждую неделю
- Комиссия до 5 лет за каждого referral

Улучшение сократительной функции сердца при помощи ингибирования NF-?B у утрофин/дистрофин дефицитных мышей
Dawn A Delfin,#1 Ying Xu,#2 Jennifer M Peterson,#3 Denis C Guttridge,#3 Jill A Rafael-Fortney,#1 and Paul ML Janssen #2
#2
1Department of Molecular and Cellular Biochemistry, Columbus, OH, USA
2Department of Physiology and Cell Biology, Columbus, OH, USA
3Department of Molecular Virology, Immunology, and Medical Genetics, The Ohio State University, Columbus, OH, USA
 Corresponding author.
Corresponding author.
#Contributed equally.
Dawn A Delfin: *****@***edu; Ying Xu: *****@***osu. edu; Jennifer M Peterson: Jennifer. *****@***edu; Denis C Guttridge: Denis. *****@***edu; Jill A Rafael-Fortney: *****@***edu; Paul ML Janssen: *****@***edu
Received February 3, 2011; Accepted May 17, 2011.
This is an Open Access article distributed under the terms of the Creative Commons Attribution License (http://creativecommons. org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Предпосылки
Миодистрофия Дюшена - это дегенеративное заболевание поперечно-полосатых мышц, вызванное отсутствием дистрофина. [1]. Несмотря на потерю силы в мышцах конечностей и потерю способности к передвижению, которые являются первыми признаками болезни, пациенты с миодистрофией Дюшена обычно умирают от сердечной или дыхательной недостаточности. У 95% пациентов развивается дилатационная кардиомиопатия, и около 25 % пациентов умирает от сердечной недостаточности. [2]. Эти числа обуславливают разработку методов лечения, направленных на улучшении дыхательных функций.[3].Это предположение лучше работает у пациентов с миодистрофией Беккера, у которых мутации в дистрофине вызывают более мягкое повреждение скелетных мышц, но для которых характерно развитие сердечной недостаточности. [3].
Улучшение функции скелетных мышц является главным фокусом для разработки лечения миодистрофии Дюшена и Беккера. Однако нацеленность только на скелетные мышцы может потенциально ухудшать существующую сердечную дисфункцию[4].Для того чтобы улучшить качество и продолжительность жизни, необходимо предупреждение и ли лечение прогрессирующего уменьшения сократительной функции сердца. Наши недавние исследования показали, что ингибирование NF-?B может улучшать функцию мышц конечностей и диафрагмы у дистрофин - дефицитных mdx мышей[5,6].Этот эффект достигнут при помощи небольшого пептида из 11аминокислот, названного NBD (NEMO Binding Domain), который связывается с С-терминальным участком IKK? и IKK? каталитических компонентов I?B киназы (IKK) предупреждая ассоциацию с NF-?B эсенциальным модулятором (NEMO)регуляторной субединицы. NBD снижает уровень сигналов NF-?B, уменьшая воспаление, улучшая регенерацию миофибрилл и сократительную функцию диафрагмы у mdx мышей[5,6].
Интересно отметить, что из всех фармакологических ингибиторов, проверенных на усовершенствование скелетных мышц на моделях миодистрофии Дюшена у животных, ни один, к нашему вниманию, не был непосредственно проверен на способность улучшить сердечную функцию. Недавние исследования даже предполагают, что текущий стандарт фармакологического лечения миодистрофии Дюшена кортикостероидами ухудшает сердечную функцию у mdx модели [7,8].
Не известно, может ли сердечная сократительная функция быть улучшена лечением NBD, но оно показало свою способность уменьшать воспалительные реакции и стимулировать новый рост скелетных мышц, приводящий к улучшенной сократительной функции, проверяя потенциал NBD по улучшению сердечной функции у модели кардиомиопатии при миодистрофии Дюшена, результат гарантирован. С этой целью, мы сосредоточили наше текущее исследование на основном открытии эффективной блокады NF-? B для улучшенния сердечной сократительной функции. Мы использовали для модели кардиомиопатии при миодистрофии Дюшена dko мышей, у которых отсутствует как дистрофин, так и компенсирующий его недостаток утрофин [9], сначала мы установили, что у 8 недельных мышей сократительная функция сердца существенно нарушена [10], и имеются классические патофизиологические маркеры сердечной недостаточности со снижением сократительной функции и отрицательным соотношением сила/частота[11], и сильным снижением ?-адренэргического ответа[12].В дополнение этот тип мышей показал дегенерацию сердечных мышц, и в возрасте 10 недель мы обнаружили замену поврежденных кардиомиоцитов на фиброзную ткань [13], сходные процессы наблюдаются у пациентов с миодистрофией Дюшена [14] и у большинства людей с сердечной недостаточностью [15,16]. Поэтому усовершенствование сердечной функции у этих мышей имеет возможное терапевтическое значение не только для кардиомиопатии при мышечных дистрофиях, но также и возможно для более многочисленного населения пациентов, страдающих от сердечной сократительной дисфункции.
В этом исследовании, чтобы полностью оценить функциональные аспекты лечения NBD, мы исследовали и сократительную функцию миокарда и ее регулирование у dko мышей. Мы оценили зависимую от длины активацию, зависимую от частоты активацию, и ? - адренергическое возбуждение в изолированных dko сердечных папиллярных мышцах после лечения пептидом NBD или в контрольной группе. Результаты указывают, что NBD может значительно улучшить сердечную сократительную дисфункцию у этой модели кардиомиопатии при миодистрофии.
Методы
Мыши
Утрофин/дистрофин дефицитные мыши были рождены приблизительно от 1:4 отношения пар utrn+/-;mdx мышей. Потомство было генотипировано сразу после рождения [9] и разделены на группы. Эксперементальный протокол был одобрен комитетом по работе с животными в университете Огайо. Синтез пептида
Синтез пептида NBD описан прежде[5].
Режим лечения
Лечение NBD начиналось, когда мышам не было еще и одной недели. NBD был разведен в 10% DMSO фосфатном буфере (PBS) и введен при помощи интраперитонеальной инъекции 3 раза в неделю пока мыши не достигли возраста 8 недель. Поскольку мыши активно росли во время лечения, перед каждой инъекцией до 4 недель, затем один раз в неделю, вводилось 10 мг/кг пептида. В предидущих эксперементах применение пептида не показало функциональных различий с контрольной группой. [5,17].Контрольная группа состояла из dko мышей, получавших те же компоненты в равном объеме без пептида (10% DMSO in PBS).
EMSA и вестерн блот
EMSA и вестерн блот проведены как описано прежде для мышечной ткани [5,6,18] сердца из всех групп мышей. Используя экстрактивный буфер с ингибиторами протеаз сердечная ткань гомогенизирована. После инкубации и центрифугирования ядерные экстракты изолированы с использованием буфера и стандартных ингибиторов протеаз. Ядра растворены и перемешаны и использованы в EMSA анализе. Эти нуклеарные экстракты были инкубированы с радиоактивными олигонуклеатидами, содержащими NF-?B сайт и фракционированы в 5% полиакриламидном геле (EMSA) или использованы в вестерн блот анализе.
Оценка физиологии сокращения
В конце режима лечения сократительная функция сердечной мышечной ткани была оценена in vitro, как описано прежде [10,19,20]. Вкраце, под глубоким наркозом B сердца были быстро удалены и помещены в раствор Кребса-Хенселента. Правый желудочек был вскрыт и папиллярные мышцы были иссечены под стереомикроскопом. Мышцы былиустановленны в эксперементальной камере с раствором Кребса-Хенселет, содержащий 1.5 мМ Ca2+, при 37°C. Мышцы были стимулированы электрическим током для стимуляции сокращений. Во первых после уравновешивания мышц в установке, мышечная длина была увеличена до возможных пределов. Эта длина считалась оптимальной. Поскольку сердце регулирует силу сокращения при помощи нескольких механизмов, важно не только оценить основныепараметры сокращения, но и ответ на нормальные физиологические регуляторные механизмы.
Мы оценили главные три механизма регуляции силы сокращения: зависимым от длины механизм, сила-зависимый механизм и ?-адренэргическая стимуляция. После оценки основных параметров сокращения, была произведена оценка стимуляции силой 4Гц у каждой мышцы, как описано прежде. [10,19]. Гистология
После физиологического анализа образцы сердечных мышц были удалены Остальная мышечная ткань была заморожена при оптимальной температуре разрезания в охлажденном жидким азотом изопентане.. Серия секций (8нм) были сделаны из блоков мышечной ткани. Для изучения гистологии секции фиксированы 100% этанолом и окрашены гематоксилином, эозином. Иммунофлюоресценция была проведена для выявления участков фиброза и иммунных клеток в участках повреждения. Не фиксированные секции были помещены в KPBS (16.4 мМ K2HPO4, 3.6 мМ KH2PO4, 160 мМ NaCl) на 5 минут, затем помещен в KPBS + 1% желатин на 15 минут. Образцы были отмыты в KPBS + 0.2% желатине (KPBSG), затем инкубированы в течение 2 часов с антителами против коллагена1, которые были растворены в KPBSG + . 1%нормальной козлиной сыворотке. (Abcam, Cambridge, MA, ab292 rabbit polyclonal) в разведении 1:200, ER-TR7 (Abcam ab51824 rat monoclonal) в разведении 1:100, или CD45 (BD Pharmingen, Franklin Lakes, NJ, 550539 rat monoclonal) в разведении 1:50. Один час с Образцы были отмыты и потом инкубированы Cy3-конъюгированными козлиными вторичными антителами против кроличьих иммуноглобулинов IgG (Jackson Immuno Research, West Grove, PA,111-165-144) или крысиных IgG (Jackson Immuno Research 712-165-153), разведенных 1:100 в KPBSG + 1% нормальной козлиной сыворотке для обнаружения связанных антител. Образцы были опять промыты и затем установлены в Vectashield (Vector Labs, Burlingame, CA)

c 2 мг/мл DAPI (Sigma, Saint Louis, MO) для окраски ядер. Флюоресценция наблюдалась при помощи Nikon Eclipse 800 микроскопа (Nikon Corporation, Tokyo, Japan) изображения получены с помощью SPOT-RTslider камеры и программного обеспечения SPOT (Diagnostic Instruments, Inc., Sterling Heights, MI). В контрольных эксперементах использовали вторичные антитела.
Статистика
Сила сокращения анализирована с использованием t-теста или ANOVA. Значимое значение P < 0.05
Результаты
Через 8 недель после лечения была произведена оценка функциональных и гистологических параметров сердечной ткани мышей. Сила сокращения изолированных сердечных мышц была оценена первой. Эти линейные мышцы содержали кардиомиоциты, фибробласты, эндотелиальные клетки и устроены линейно для облегчения оценки качественных и количественных показателей механической функции и регуляторных процессов. [21,22]. В основных условиях (оптимальная длина, 4 Гц стимуляция, 37°C), активная сила развивалась в мышцах NBD леченных dko мышей значительно выше, чем в контрольной группе. (12.5 ± 1.8 vs. 5.2 ± 1.8 mN/mm2, P < 0.05, Рисунок 1А ). По количеству эти различия схожи наблюдаемым в предидущих исследованиях дикого типа (WT) мышей и dko мышей [10], включая полное восстановление силы при помощи NBD. Диастолическое напряжение нуждается в достижении оптимального активного напряжения значительно отличающегося у двух групп и были 11.7 ± 1.9 mN/mm2 в контрольной группе и 10.8 ± 1.9 mN/mm2 в группе получавшей NBD (P = 0.75). Максимальная скорость сокращения и расслабления (dF/dtmax и dF/dtmin соответственно) была также значительно выше в мускулах от мышей получавших NBD (P <0.05, Рисунок 1B). Однако, увеличение производной силы - главным образом, следствие полного увеличения силы. Когда мы оценили время возбуждения для достижения максимума напряжения, и время от пиковой напряжения до 90%-ого расслабления, мы только наблюдали маленькое, незначительное ускорение сократительной кинетики (рисунок 1C). Это также указывает на усовершенствование функции, поскольку часто развитие силы увеличения по существу приводит к замедлению расслабления возможно ослабляя диастолическую функцию [23] Ясно что, несмотря на увеличенную силу в мускулах от NBD леченных мышей, кинетика расслабления не была медленнее, и даже имела тенденцию быть быстрее.
| Рисунок 1 |
Чтобы оценить, было ли развитие силы увеличено независимо от его регулирующих механизмов, мы исследовали были ли нормальные физиологические регулирующие механизмы, которые увеличивают сердечную сокращаемость, изменены лечением NBD. Нормальное физиологическое регулирование сократительной функции происходит через несколько механизмов, и используется, чтобы увеличить кровоток, когда физическая потребность выше, той, которая наблюдается при тренировках. Самым известным из этих регулирующих механизмов является механизм Франка-Старлинга, который приводит к увеличению сократительной силы, когда увеличена предварительная нагрузка (желудочковый объем в начале сокращения) или длина сердечных мышечных клеток. Чтобы подражать этому механизму в нашем эксперементе, мы оценили сократительную силу при 4 различных длинах мышцы (представляющий различное состояние желудочка), в пределах от 85 % оптимальной длины до оптимальной длины. Мы заметили, что зависимая от длины активация по существу (форма кривой) не отличалась в мышцах от мышей получавших NBD, которые рассматривают по сравнению с контрольными dko мышами (Рисунок 2). Поэтому, поскольку длина мускула увеличилась, сила сокращения увеличилась в обеих группах. Статистический анализ через АНОВУ указал, что различие в силе было существенным, как результат длины, но не взаимодействия между этими двумя параметрами, указывая, что зависимое от длины поведение неизменно после лечения NBD.
| Рисунок 2 |

Затем, мы исследовали эффект лечения NBD на втором механизме регулирования сердечного сокращения: зависимое от частоты поведение. От основных условий при оптимальной длине частота возбуждения была увеличена от 4 до 6, 8, 10, 12 и 14 гц, охватывая в естественных условиях диапазон характерный для мышей (24). Как мы ранее показали, у контрольных dko мышей отмечалась патологическое отрицательное отношение частоты к силе с увеличением уровня возбуждения, приводящего к уменьшению пиковой сократительной силе. АНОВА указала не только, что и частота и лечение были существенными, но также и что взаимодействие существенно отличалось между NBD и контролем dko мышей. Благодаря распространению в абсолютных силах, это трудно иллюстрировать (Рисунок 3A) но то, когда каждый мускул нормализован к его собственному начальному уровню силы в 4 гц, эти отношения, более легко представить (Рисунок 3B). У рассматриваемых мышей контрольной группы изменение частоты возбуждения с 4 до 10 гц привело к 46 ± 6%-ых потерям силы (p <0.05, отрицательной частоты

силы). Напротив, у NBD-леченных мышей изменение в силе от частоты возбуждения на 4 до 10 гц не было существенным. Эти плоские отношения частоты силы снова почти идентичны по качеству и количеству по

сравнению с результатами, полученными у здоровых WT мышей (10). Таким образом, лечение NBD значительно предотвратило зависимое от частоты ухудшение поведения мышц. Когда уровень возбуждения увеличился, обе группы ответили фактически равным увеличением уровня кинетики. Среднее ускорение 50%-ого времени расслабления составляло 10.2 миллисекунд у рассматриваемых мышей, получавших NBD, против 9.8 миллисекунд у рассматриваемых dko мышей контрольной группы.
| Рисунок 3 |

Третий главный механизм, который регулирует сократительную функцию в естественных условиях, является ? - адренергическое возбуждение. Чтобы оценить этот ответ, мы обработали сокращающиеся мышцы увеличивающимся концентрациям ? – адренергического агониста изопротеренола. Как показано на рисунке 4,4, ответ у контрольной группы к изопротеренолу патологически слаб, со средним увеличением силы только 2.8 mN/mm2. Этот слабый ответ находится в соответствии с нашими ранее изданными результатами(10), ответ у рассматриваемых dko мышей, получавших NBD, является здоровым и в более чем три раза превышает (среднее число 10.0 mN/mm2) ответ, наблюдаемый в контрольной группе. Снова, этот восстановленный ответ был подобен по величине здоровым мышам дикого типа в нашем предыдущем исследовании. Ускорение расслабления было подобно в обеих группах, и не существенно отличалось.
| Рисунок 4 |

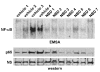
Затем, мы исследовали фармакодинамическую эффективность пептида NBD в сердечных мышцах рассматриваемых мышей dko. Во всех группах NF-? B связывающая активность ДНК, так же как ядерные уровни p65 субъединицы NF-? B были высоки в dko сердце. Вообще, эта активация была эффективно уменьшена у dko мышей получавших NBD (Рисунок 5). Эти результаты были сравнимы с нашими предыдущими результатами в мышцах диафрагмы mdx мышей, получавших NBD, вместе с тем подтверждая, что NBD улучшает сердечную сократительную дисфункции у dko мышей посредствам блокады сигналов NF-? B.
| Рисунок 5 |
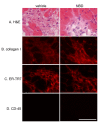
Наконец, мы проверяли привело ли лечение NBD dko мышей к улучшению сердечных гистологических особенностей этой модели. В возрасте между восемью и десятью неделями у dko мышей обнаруживается миокардиальное повреждение, сопровождаемое фиброзом в поврежденных регионах [13]. Несмотря на улучшение сократительной функции, наблюдаемой после лечения NBD, и подтвержденной роли NF-? B в воспламенении, гистологические особенности dko миокарда не были заметно улучшены применением NBD.
Мы наблюдали большие участки фиброза (Рисунок 6A) в сердцах большинства dko мышей в этом исследовании независимо от лечения (5 из 7 [71%-ых] NBD, против 3 из 4 [75%-ых] в контрольной группе). Количество повреждения сердечной ткани в обеих группах dko мышей в этом исследовании было сопоставимо с повреждением, существующим у десяти dko мышей недельного возраста [13]. Иммунофлюоресценция с использованием антител к коллагену I показали, что фиброзные области содержали чрезвычайно много колллагена в обеих группах (Рисунок 6B). Фибробласты, которые, как известно, были ответственны за большую часть сердечной модернизации при кардиомиопатии [15], присутствуют в больших количествах и в NBD и в контрольных dko сердцах в областях фиброза (Рисунок 6C). Инфильтраты иммунных клеток, вероятно, требуются для поврежденния миокардиальной ткани, но в момент времени проанализированном здесь, мы не могли обнаружить присутствие большого количества этих клеток, кроме спорадических клеток гемопоэтического происхождения в поврежденных областях сердец или при лечении NBD или в контрольной группе, обнаруженных при помощи антител против CD 45 (Рисунок 6D) или CD 11b или более определенных маркеров F4/80 макрофагов (данные не показаны).
.
| Рисунок 6 |
Дискуссия
Сердечная сократительная дисфункция - одна из главных причин смерти при миодистрофии Дюшена. Клиническое лечение этого аспекта МДД является главным для увеличения продолжительности и качества жизни. В этом исследовании мы показали, что пептид, называемый NBD, который блокирует NF-? B передачу сигналов, может восстановить сердечную сократительную дисфункцию у мышей - моделей МДД. Мало того, что лечение NBD существенно увеличивало силу сокращения, а также улучшило ключевые управляющие механизмы силы сокращения, которые как правило ослабевают у пациентов с сердечной недостаточностью, включая поведение частоты/силы и ответ на ? - адренергическое возбуждение [11,12,25].
Для этого исследования мы не включали дополнительные модели мышечной дистрофии или мышей дикого типа. Однако, мы можем сравнить ответ с данными нашего предыдущего исследования [10], в котором мы использовали здоровые мыши дикого типа так же как mdx мыши. Мыши Mdx - генотипная, часто используемая модель миодистрофии Дюшена с намного более умеренным фенотипом (меньше сократительная дисфункция) по сравнению с dko мышами. В нашем текущем исследовании мы использовали маленькие папиллярные мышцы правого желудочка со средней шириной 266 ± 8 мкм, 177 ± 5 мкм в центре, и 1.04 ± 0.08 мм длиной. В нашей предыдущей работе [10], мы использовали желудочковые тонкие трабекулы от mdx, dko мыши, и C57Bl/10 группу контроля. Трабекулы, используемые ранее, были немного более узкими (средняя ширина 220 мкм) и дольше (средние 1.5 мм). Однако, хотя трабекулы очень хорошо удовлетворяют для оценки сократительной функции вообще [26], их частота менее предсказуема чем у папиллярных мышц. Когда мы нормализуем оба исследования сократительная сила dko мышей, показанная на рисунке7, мы можем установить, что увеличение сократительной силы является очень существенным. Фактически, силы, произведенные во время основных условий у рассматриваемых dko мышей, получавших NBD, относительно подобны полученным у мышей дикого типа C57, и выше чем полученные у не леченных mdx мышей. Кроме того, ответы на увеличенную частоту возбуждения так же как на ? - адренергическое возбуждение после лечения NBD dko мышей, и приближается к параметрам у здоровых мышей дикого типа C57 [10].
| Рисунок 7 |

Увеличенная сократительная сила была вероятна не непосредственным влиянием измененной гистологии миокарда. Мы не наблюдали значительного сокращения фиброза в dko миокарде после лечения с NBD. Однако, мы не можем исключать того, что местное усовершенствования гистологии папиллярных мускулов могут играть роль. Большая часть области правого желудочка и перегородки, где мускулы были вырезаны, является неподходящей для гистологического анализа. Мышцы, используемые для физиологических измерений силы, после экспериментирования, являются также не подходящими для гистологического анализа и последующего коррелативного анализа. Таким образом, мы не можем показать потенциальное гистологическое изменение в препаратах, где функция была фактически оценена. Однако, учитывая широко распространенный фиброз, который все еще присутствовал в остающейся желудочковой ткани после лечения NBD, местное улучшение гистопатологии, являющейся прежде всего ответственный за улучшенную функцию, довольно маловероятно. В настоящее время вне области этого исследования, мы можем только размышлять об основных молекулярных событиях, которые в конечном счете приводят к улучшению сократительной функции.
Первопричина ослабленной сократительной функции сердца в терминальной стадии сердечной недостаточности часто независима от происходящей причины остановки сердца у модели животных или пациенте. Лечение кальцием, которой ослабляет, - центральное открытие при остановке сердца в терминальной стадии. В отказе человеческого сердца нормальная положительная частотная характеристика силы как правило строго притупляется, или даже становится отрицательной, и является признаком фенотипичной дисфункции (11, 24, 25) У нормальных, здоровых мышей, когда частота сокращения увеличена, развитие силы мышц вообще немного увеличено [27] или по крайней мере не уменьшается, в то время как расслабление всегда быстрее [19]. Однако, у мышей с сердечной дисфункцией, таких как dko мышь, используемая в этом исследовании, отношения частоты силы отрицательны [10]. Мы

показываем, что лечение NBD не только увеличивает сократительную силу dko миокарда, но и это также значительно улучшило отношения частоты/силы. Этот ответ больше не был в значительной степени отрицателен, и даже вернулся к положительному на более низком уровне частотного диапазона, напоминая зависимое от частоты поведение, как правило найденное у здоровых мышей. Восстановление нормальной частотной характеристики силы - таким образом косвенная улика, что улучшение поступления кальция может быть главным основным фактором в функциональном усовершенствовании dko миокарда после лечения NBD.
NF-? B и ионы кальция являются и многогранными сигнальными молекулами и взаимодействиями между концентрацией иона кальция, и NF-? B были зарегистрированы. Например, в гладких мышцах, NF-? B отрицательно воздействуют на каналы кальция [28], и таким образом лечение NBD могло потенциально привести к улучшению регуляции этих каналов кальция, улучшая функцию, облегчая приток кальция. Кроме того, блокада NF-? B, как показывали, была в состоянии облегчить саркоплазматическое напряжение, и взаимодействовать с уровнями саркоплазматической/эндоплазматической кальциевой АТФазы (SERCA), которая ответственена за перенос ионов кальция в цитоплазме [29]. NF-? B в скелетной мышце, как показывали, смодулировал экспрессию изоформ NO-синтазы [30], которые играют важную роль в поддержании сердечно-сосудистого гомеостаза, главным образом, через регуляцию кальция. Наконец, недавний отчет Панамы и коллег [31] показали что NF-? B регулирует переходный поток калия направленный наружу в сердце, далее представляющие свидетельства для роли NF-? B регулированнии процессов сокращения - возбуждения (EC). Поэтому, хотя у нас нет никакого прямого неопровержимого доказательства на данном этапе, NBD может улучшить сократительную функцию в dko миокарде через улучшение EC/кальций, а не через предотвращение гистологически обнаружимого повреждения. Дистрофичная скелетная функция мышц может быть улучшена низкими уровнями дистрофина в отсутствие гистопатологического улучшения [32]. Поэтому, подобное усовершенствование функции нефиброзного дистрофичного миокарда может составлять результаты нашего исследования. Далее предназначенные исследования обязаны объяснять возможные механизмы и могли включать электрофизиологические и гемодинамические оценки [33, 34], так же как внутриклеточная обработка кальция [19]. Любая терапевтическая стратегия, вовлекающая NBD, может потребовать комбинированого подхода с фактором, который предотвратил бы сердечное повреждение
В дополнение к уменьшенной сокращаемости и отрицательной частотной характеристике силы, известно, что оба у пациентов с сердечной недостаточностью, так же как во многих моделях животных сердечной дисфункции, физиологического ответа на ? - адренергическое возбуждение строго притуплены [12]. В невылеченном dko миокарде это притупляло ? - адренергический ответ [10], и в данном исследовании мы нашли, что лечение NBD значительно улучшает этот ответ. Главные основные мероприятия молекулярного уровня, которые приводят к увеличенной сокращаемости после ? - адренергического возбуждения, могут быть в повышении внутриклеточных процессов перемещения кальция. Таким образом, тот же самый механизм, ответственный за улучшенную частотную характеристику силы, мог быть основным фактором для усовершенствования этого ? - ответа.
Ссылки
Blake DJ, Weir A, Newey SE, Davies KE. Function and genetics of dystrophin and dystrophin-related proteins in muscle. Physiol Rev. 2002;82:291–329. [PubMed] McNally EM. New approaches in the therapy of cardiomyopathy in muscular dystrophy. Annual Review of Medicine. 2007;58:75–88. doi: 10.1146/annurev. med.58.011706.144703. [PubMed] [Cross Ref] Duan D. Challenges and opportunities in dystrophin-deficient cardiomyopathy gene therapy. Hum Mol Genet. 2006;15(Spec No 2):R253–261. [PMC free article] [PubMed] Townsend D, Yasuda S, Li S, Chamberlain JS, Metzger JM. Emergent dilated cardiomyopathy caused by targeted repair of dystrophic skeletal muscle. Mol Ther. 2008;16:832–835. doi: 10.1038/mt.2008.52. [PMC free article] [PubMed] [Cross Ref] Acharyya S, Villalta SA, Bakkar N, Bupha-Intr T, Janssen PML, Carathers M, Li ZW, Beg AA, Ghosh S, Sahenk Z, Weinstein M, Gardner KL, Rafael-Fortney JA, Karin M, Tidball JG, Baldwin AS, Guttridge DC. Interplay of IKK/NF-kappaB signaling in macrophages and myofibers promotes muscle degeneration in Duchenne muscular dystrophy. J Clin Invest. 2007;117:889–901. doi: 10.1172/JCI30556. [PMC free article] [PubMed] [Cross Ref] Peterson JM, Kline W, Canan BD, Ricca DJ, Kaspar BK, Delfin DA, DiRienzo K, Clemens PR, Robbins PD, Baldwin AS, Flood P, Kaumaya P, Freitas M, Kornegay JN, Mendell JR, Rafael-Fortney JA, Guttridge DC, Janssen PML. Peptide-based inhibition of NF-?B rescues diaphragm muscle contractile dysfunction in a murine model of Duchenne muscular dystrophy. Molecular Medicine. 2011. in press. Guerron AD, Rawat R, Sali A, Spurney CF, Pistilli E, Cha HJ, Pandey GS, Gernapudi R, Francia D, Farajian V, Escolar DM, Bossi L, Becker M, Zerr P, de la Porte S, Gordish-Dressman H, Partridge T, Hoffman EP, Nagaraju K. Functional and molecular effects of arginine butyrate and prednisone on muscle and heart in the mdx mouse model of Duchenne Muscular Dystrophy. PLoS One. 2010;5:e11220. doi:

Переведено проектом МОЙМИО http://www. mymio. org
Оригинальная статья http://www. ncbi. nlm. nih. gov/pmc/articles/PMC3212940/?tool=pubmed

