

Партнерка на США и Канаду по недвижимости, выплаты в крипто
- 30% recurring commission
- Выплаты в USDT
- Вывод каждую неделю
- Комиссия до 5 лет за каждого referral
ГБОУ ЛГК на Юго-Востоке ("Московский Химический Лицей 1303")
Институт органической химии им. РАН
Разработка методов получения силил - и борозамещенных гем?фторгалогенциклопропанов и исследование их превращений с раскрытием цикла
Исследовательская работа
Выполнили:
Научный руководитель:
Москва – 2017
ОГЛАВЛЕНИЕ
1. Введение.....................................................................................................................................4
2. Получение фтораллилсиланов и фтораллилборанов (обзор литературы)…………...……5
2.1. Силилирование аллильных нуклеофилов……………………………………...…..5
2.2. Реакции нуклеофильного замещения аллильного галогена……………..……….6
2.3. Реакции алкенилирования ?-галогенсиланов и боранов…………………………8
2.4. Реакции алкенилирования ?-силилзамещенных металлорганических реагентов……………………………………………………………………………………….8
2.5. Прочие методы……………………………………………………………………9
3.Результаты и обсуждение........................................................................................................10
3.1. Получение гем-фторбромциклопропанов..............................................................10
3.2. Изомеризация гем-фторгалогенциклопропанов, катализируемая соединениями меди...............................................................................................................................................13
4. Экспериментальная часть.......................................................................................................16
4.1 Синтез силилзамещенных гем-фторбромциклопропанов....................................16
4.2 Циклопропанирование под действием CBr2FCO2Na............................................18
4.2.1. Синтез дибромфторацетата натрия..........................................................18
4.2.2. Оптимизация условий циклопропанирования........................................18
4.2.3. Циклопропанирование (1-фенилэтенил)пинаколбороната……………19
4.3 Изомеризация силилзамещенных гем-фторбромциклопропанов......................19
5.Выводы......................................................................................................................................21
6.Литература.................................................................................................................................22
Список сокращений:
ГХ – газовая хромотография
КССВ – константа спин-спинового взаимодействия
ТЭБАХ – триэтилбензиламмоний хлорид
циклопроп. – циклопропан
хч – химически чистый
экв. – эквивалент
ЯМР – ядерный магнитный резонанс
Bpin - пинаколборонил
DMPU - 1,3-диметил-3,4,5,6-тетрагидро-2(1H)-пиримидинон
DMSO – диметилсульфоксид
HMPA – гексаметилфосфорамид
TBAF – тетрабутиламмоний фторид
pin – пинаколил
p-Tol – пара-толил
THF – тетрагидрофуран
Список сокращений для экспериментальной части:
д. - дублет
д. д. – дублет дублетов
м. – мультиплет
с. – синглет
т. - триплет
уш. - уширенный
1. Введение
Фторорганические соединения широко используются в современной медицинской химии для разработки новых лекарственных препаратов, благодаря возможности фтора оказывать уникальное влияние на физические, химические и биологические свойства органических структур: увеличивать проницаемость через мембраны клеток, метаболическую стабильность, биологическую активность и липофильность в биологических системах при введении атома фтора в структуру молекул [1–5]. Фтор участвует во многих биохимических реакциях, регулирует активность ряда ферментов, таких как аденилатциклаза, липазы, эстеразы, лактатдегидроденазы и т. п. Как следствие, в последние годы стремительно растет процент лекарственных препаратов (в настоящее время их количество достигает 40%), содержащих в своей структуре один или несколько атомов фтора. Для получения подобных структур перспективными синтонами являются фтораллилбораны и фтораллилсиланы из-за своей способности вступать в разнообразные реакции аллилирования с высокой диастерео - и энантиоселективностью [6-8].
В то же время, фтораллилбораны и фтораллилсиланы являются малоизученными классами соединений. Связь С-B, как и связь С-Si поляризованы в направлении углерода за счет его большей электроотрицательности, что с одной стороны делает эти соединения хорошими C-нуклеофилами, а с другой — приводит трудностям при получении таких структур из-за близости атомов фтора к нуклеофильному центру, а также к их высокой неустойчивости и склонности к побочным процессам дефторирования.
Целью данной работы является разработка метода получения фтораллилсиланов и фтораллилборанов, основанного на процессах раскрытия цикла по типу циклопропил-аллильной трансформации силил - и борозамещенных циклопропанов, получаемых карбенным циклопропанированием соответствующих олефинов.
2. Получение фтораллилсиланов и фтораллилборанов (обзор литературы)
Целью настоящего обзора является анализ различных методов синтеза фтораллилсиланов и фтораллилборанов.
2.1. Силилирование аллильных нуклеофилов
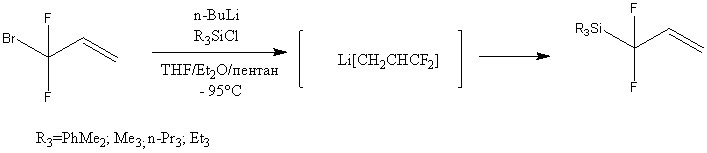
Был разработан метод получения 1,1-дифтораллилсиланов в тетрагидрофуране, пентане и диэтиловом эфире (THF/пентан/Et2O=5:1:1) под действием n-BuLi при температуре -95оС. Реакция протекает через образование Li[CH2CH=CF2]. В данных условиях были получены различные (1,1-дифтораллил)триалкилсиланы с довольно хорошими выходами (51-74%).
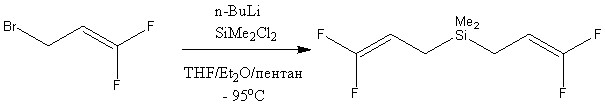
Ранее было рассмотрено получение различных 1,1-дифтораллилсиланов с генерированием Li[CH2CHCF2] из Me3SnCH2CH=CF2 в аналогичных условиях. Однако же отмечалось медленное прохождение реакций и образование отличающихся продуктов.[9-10]
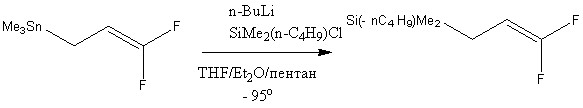
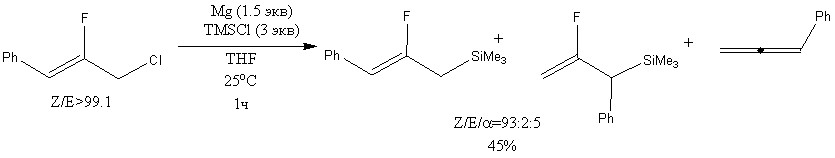
Силилирование (Z)-2-фтор-3-фенилаллилхлорида под действием TMSCl в присутствие Mg дает смесь фтораллилсиланов и фенилаллена при комнатной температуре. Были получены Z-изомер с выходом 45%. При силилировании смеси аллилхлоридов была получена смесь изомеров с выходом 50%.[11]
2.2. Реакции нуклеофильного замещения аллильного галогена
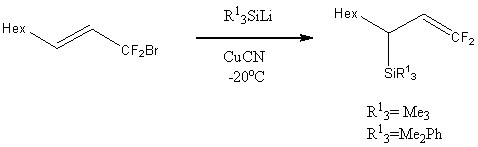
Под действием Me3SiLi в HMPA и Et2O в присутствии цианида одновалентной меди при -20оС из 1-бром-1-фторнонена были получены 1,1-дифтор-3-триметилсилил-l-нонен и 1,1-дифтор-3-(диметилфенилсилил)-1-нонен с выходами 78% и 75%, соответственно.[12]
При использовании системы дисилан/TBAF(0.1 экв) в качестве источника силильного аниона были получены различные гем-дифтораллилсиланы аллильным замещением атома фтора в 3,3,3-трифторпропенах с выходами от 56% до 85% (таблица 1). Реакция протекает как в полярных апротонных растворителях (DMPU или HMPA), так и в менее полярном THF.[13]
Таблица 1. Получение дифтортриалкилсиланов в TBAF | ||||
Исходный олефин | Условия | Продукт | Выход, (%) | |
1 | CF3-CH=CH2 | PhMe2SiSiMe2Ph/TBAF/DMPU | CF2=CHCH2SiMe2Ph | 84 |
2 | CF3-CH=CH2 | PhMe2SiSiMe2Ph/TBAF/HMPA | CF2=CHCH2SiMe2Ph | 85 |
3 | CF3-CH=CH2 | Me3SiSiMe2Ph/TBAF/THF | CF2=CHCH2SiMe2Ph | 85 |
4 | CF3-CH=CH2 | Me3SiSiMe2Ph/TBAF/DMPU | CF2=CHCH2SiMe2Ph | 66 |
5 | CF3-CH=CH2 | Me3SiSiMe2Ph/TBAF/HMPA | CF2=CHCH2SiMe2Ph | 64 |
6 | CF3-CH=CH2 | PhMe2SiLi/THF | CF2=CHCH2SiMe2Ph | 62 |
7 | CF3-CH=CH2 | Me3SiSiPh3/TBAF/HMPA | CF2=CHCH2SiPh3 | 56 |
8 | CF3-C(Me)=CH2 | PhMe2SiSiMe2Ph/TBAF/THF | CF2=C(Me)CH2SiMe2Ph | 68 |
9 | CF3-C(Me)=CH2 | Me3SiSiMe2Ph/TBAF/HMPA | CF2=C(Me)CH2SiMe2Ph | 74 |
10 | CF3-C(Me)=CH2 | PhMe2SiLi/THF | CF2=C(Me)CH2SiMe2Ph | 71 |
Не было получено хороших результатов в условиях 5 и 10 для THF, поэтому был использован образованный под действием n-BuLi PhMe2SiLi. Начиная с 3, всегда получались диметилфенилсилильные производные, как продукты, вследствие селективного атаки фторида на менее затрудненный атома кремния дисилана для получения диметилфенилсилил аниона.
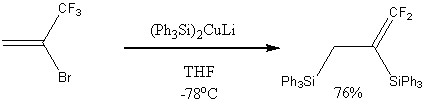
Предложен метод получения 1-дифтор-2,3-ди(трифенилсилил)пропена с 2-бром-3,3,3-трифторпропеном (2-BrTFP) в THF при –78оС под действием ди(трифенилсилил)купрата лития. [14]
Предложены условия для синтеза 1-фтор-2-фенил-1-триметилсилана из 2-фтор-3-фенилпроп-2-енилацетата (E / Z = 45 / 1) с избытком гексаметилдисилана в присутствии 1 мол.% Pd(PPh3)4 при 160оC в запаянной ампуле. Продукт был получен (Е / Z = 1 / 10) с выходом 75%. [15]

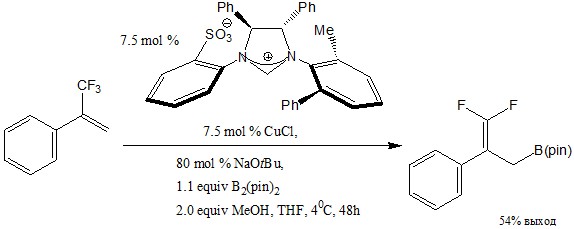
Одним из методов получения фтораллилборанов является их синтез из аллилтрифторидов и бис(пинаколато)диборана, катализируемый хиральной солью имидазолиния по методу, описанному в статье с хлоридом меди(I), трет-бутилатом натрия, метанолом в тетрагидрофуране в течение 48 часов при температуре 40С с выходом 54%.[16]
2.3. Реакции алкенилирования ?-галогенсиланов и боранов
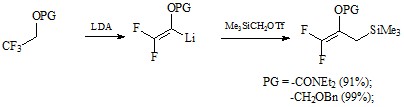
Был рассмотрен метод получения дифтораллилсилана из эфиров 2,2,2-трифторэтанола через образование дифторвиниллития и последующее замещение лития на (триметилсилил)алкильную группу с помощью Me3SiCH2OTf с выходами 91–99%.[17]
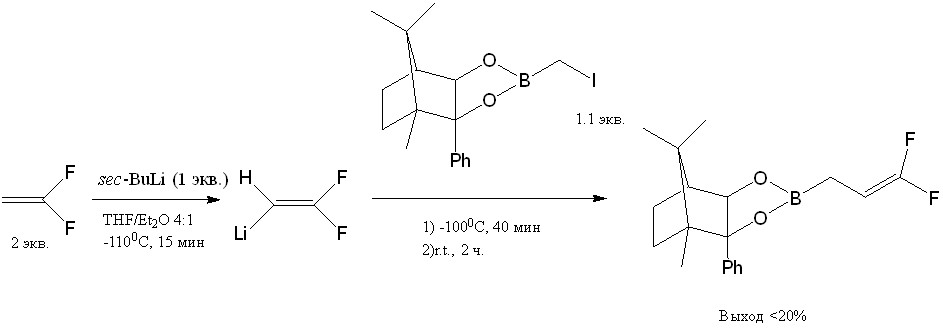
Другим возможным методом получения фтораллилборанов является их синтез с помощью дифторвиниллития из иодометилборонатов в условиях, описанных в статье [18]. Выход дифтораллилбороната в данной реакции меньше 20%.
2.4. Реакции алкенилирования ?-силилзамещенных металлорганических реагентов
Одним из возможных способов получения фтораллилсиланов является метод алкенилирования ?-силилзамещенных металлорганических реагентов.
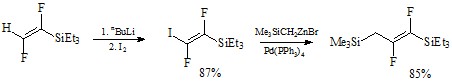
Был предложен метод получения дифтораллилсиланов из 1,2-дифтортри(этил)силилэтилена под действием BuLi и I2 и последующим замещением иода на (триметилсилил)алкильную группу с использованием Me3SiCH2ZnBr, катализируемым 3 мол.% Pd(PPh3)4 . [19]
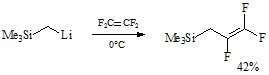
Другим возможным способом является метод замещения атома фтора в тетрафторэтилене на (триметилсилил)алкильную группу с помощью (триметилсилил)метиллития.[20]
2.5. Прочие методы
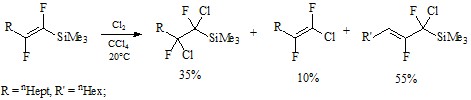
Был рассмотрен метод хлорирования дифтор(триметилсилил)этилена с получением разнообразных продуктов, в том числе 1-хлор-1,2-дифтораллилсилан с выходом 55%.[21]
Были описаны случаи получения силилзамещенного фтордиена через образование бромфторциклопропана и последующее раскрытие цикла, но атом галогена в конечной структуре также не сохранялся.[22]
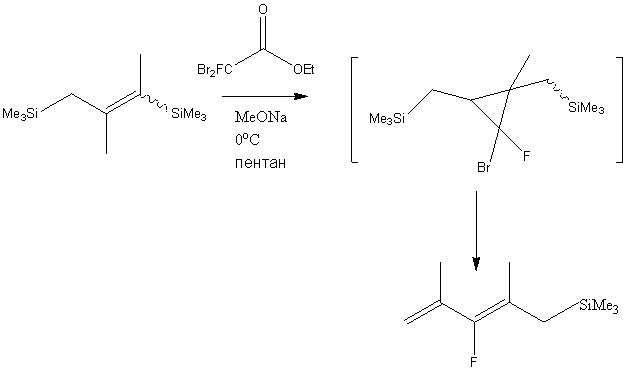
3. Результаты и обсуждение
Среди широкого многообразия методов получения фторорганических соединений важное место занимают методы, основанные на превращениях фторированных циклопропанов с раскрытием цикла, из-за широкой доступности фторциклопропанов и их способности вступать в разнообразные превращения с раскрытием цикла и сохранения атомов фтора в конечных структурах. Подход к получению фтораллилсиланов и фтораллилборанов на основе превращений фторированных циклопропанов с раскрытием цикла по типу циклопропил-аллильной трансформации представляет собой перспективную альтернативу подходам, включающим в себя различные методы прямого введения фтора в молекулу или использование других фторсодержащих синтонов [9-22]. Такие превращения протекают в условиях термолиза [23], в присутствии солей серебра (I) [24–26] или при катализе соединениями меди (I) [27–30] и приводят к получению разнообразных фтораренов, фторалкенов и фторалкадиенов.
Нами были проведены исследования, направленные на поиск новых реакционных условий и каталитических активаторов циклопропил-аллильной трансформации ранее не изученных силил - и борозамещенных гем-фторгалогенциклопропанов, изучение этих превращений в зависимости от структуры циклопропанов с целью разработки новых методов синтеза 2-фтораллилсиланов и 2-фтораллилборанов.
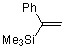
3.1 Получение гем-фторбромциклопропанов
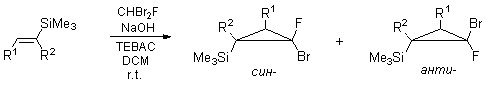
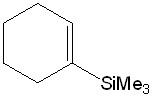
Карбенным циклопропанированием легко получаемых силильных олефинов (? - и ?-(триметилсилил)стиролов, 1?(триметилсилил)циклогексена) в условиях щелочного дегидрогалогенирования дибромфторметана [30] были получены циклопропановые структуры 1a-c (схема 1).
Фторгалогенциклопропанирование протекало с образованием смесей диастереомерных син - и анти-фторгалогенциклопропанов. Соотношение син/анти определяли методами ГХ и 19F ЯМР. Соотнесение было проведено на основании двумерных корреляционных спектров {1H-1H}-NOESY и {1H-19F}-HOESY и винициальных KCCB 1H-19F.
| ||||
№ | Исходный олефин | Продукт | Выход | син-/анти- |
1 |
|
| 52% | 30/70 |
2 |
|
| 73% | 27/73 |
3 |
|
| 21% | 77/23 |
Схема 1 |
В ходе выполнения проекта было выяснено, что в условиях генерирования фторбромкарбена щелочным дегидрогалогенированием дибромфторметана алкенилборонаты неустойчивы вследствие легкого протекания гидролиза под действием щелочи (схема 2).
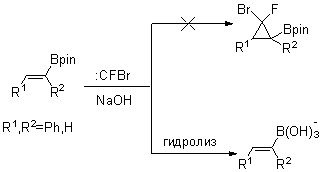
Схема 2
Были найдены условия для циклопропанирования модельного соединения (1,1- дифенилэтилена) дибромфторацетатом натрия (таблица 2), синтезированным из широкодоступной малоновой кислоты (схема 3).
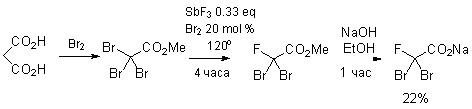
Схема 3
Для данного циклопропанирования необходимо медленное генерирование карбена для протекания реакции с исходным алкеном и избежания его димеризации или гидролиза. Вследствие этого реакцию проводили путём медленного добавления дибромфторацетата натрия к нагретому до температуры 80–120°С раствору алкена в диглиме. (схема 3.1, таблица 2)
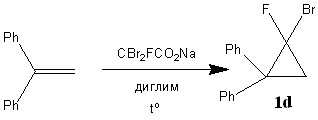
схема 3.1
Таблица 2. Отношение продукта циклопропанирования (1, 1– дифенил-2-фтор-2-бромциклопропана – 1d) к исходному 1,1-дифенилэтилену | ||
Температура нагрева | CBr2FCO2Na (экв.) | Отношение |
80o | 2 | 76:24 |
100o | 2 | 76:24 |
120o | 2 | 85:15 |
100o | 8 | 51:49 |
При использовании в качестве растворителя диоксана, никаких превращений не наблюдалось, вероятно, из-за низкой растворимости дибромфторацетата натрия. В диглиме, относительно хорошо растворяющем CBr2FCO2Na, при 80–120°C наблюдается образование циклопропана 1d. При более высоких температурах получаемый циклопропан 1d неустойчив. В таблице 2 приведена зависимость конверсии образующегося циклопропана 1d от температуры реакции и количества используемого дибромфторацетата натрия. Так, при увеличении температуры с 80°C до 100°C заметного изменения конверсии не наблюдается. Дальнейшее увеличение температуры до 120°C приводит к снижению конверсии, вероятно, из-за слишком быстрого разложения CBr2FCO2Na до фторбромкарбена. Увеличение количества используемой соли с 2 экв. до 8 позволяет значительно повысить количество образующегося 1d.
При увеличении времени нагревания реакционной смеси после добавления всего CBr2FCO2Na отношение 1d к исходному 1,1-дифенилэтилену не менялось из-за быстрого разложения CBr2FCO2Na до фторбромкарбена в условиях процесса.
Таким образом, разработанный метод был использован для получения ?-фторбром(пинаколборонил)фенил циклопропана 1e (схема 3.2).
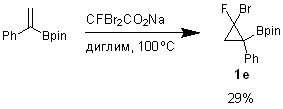
Схема 3.2
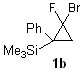
3.2. Изомеризация силилзамещенных гем-фторгалогенциклопропанов, катализируемая соединениями меди
В ходе выполнения работы были изучены различные реакционные условия для изомеризации циклопропанов 1a и 1b под действием бромида меди (I). Были использованы различные растворители и различные лиганды. (таблица 3). Обнаружено, что конверсия 1a, b более 75% наблюдалась лишь в ацетонитриле при 100°C в присутствии CuBr. В остальных случаях под действием различных лигандов раскрытия циклопропана не наблюдалось. В случаях 6-11 наблюдалось образование TMSF, процентный выход которого по данным 19F ЯМР спектроскопии относительно внутреннего стандарта отображен в таблице 3. Но следует заметить, что из-за высокой летучести TМSF (т. кип. 16°C) процентный выход нельзя считать соответствующим действительности, реальный выход TMSF, вероятно, гораздо больше.
Таблица 3. Зависимость выхода реакции изомеризации под действием бромида меди от растворителя, времени и нуклеофилов в реакции с 1a и 1b | |||||||||
№ | катализатор | Р-ль | Доп. лиганды | T,°C | t (ч) | TMSF | 1 | продукт | |
1 | 1a | CuBr 0.2 экв | MeCN | - | 100 | 5 | - | 13% | 2a |
2 | 1b | CuBr | MeCN | - | 100 | 5 | - | 98% | 2b |
3 | 1b | CuBr | MeCN | - | 100 | 8 | - | 84% | 2b |
4 | 1b | CuBr | MeCN | - | 100 | 24 | - | 34% | 2b |
5 | 1b | CuBr | MeCN | - | 100 | 48 | - | 23% | 2b |
6 | 1b | CuBr | диоксан | Морфолин | 100 | 5 | 1% | 99% | - |
7 | 1b | CuBr | диоксан | Морфолин | 100 | 8 | 1% | 95.8% | - |
8 | 1b | CuBr | DMSO | Морфолин | 100 | 5 | 28% | - | - |
9 | 1b | CuBr | DMSO | CH3COONa | 100 | 5 | 26% | 4% | - |
10 | 1b | CuBr | DMF | p-TolSO2Na | 100 | 5 | 3% | - | - |
11 | 1b | CuBr | DMSO | p-TolSO2Na | 100 | 5 | 5% | - | - |
12 | 1b | CuBr | диоксан | p-TolSO2Na | 100 | 5 | - | - | - |
При изомеризации циклопропана 1b в ацетонитриле под действием бромида меди был получен 2-фтор-3-бром-1-триметилсилил-1-фенилпропен (2b). В таблице 3 в случаях 2-5 представлена зависимость конверсии 2b от времени реакции. При изомеризации 1а в тех же реакционных условиях (случай 1) был получен 2-фтор-3-триметилсилил-3-бром-1-фенилпропен (2а). Но при увеличении времени протекания реакции для 1a конверсия не менялась, как и отношение изомерных продуктов друг к другу. Различие во времени протекания реакции изомеризации циклопропанов 1а и 1b, вероятно, обусловлено стерическими факторами в переходном состоянии [29,30].
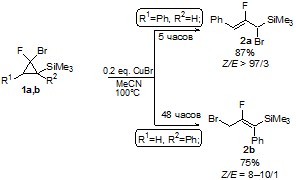
Схема 4
Полученные фтораллилсилан 2а и алкенилсилан 2b могут быть использованы для синтеза различных биологически активных структур. Введение фтораллильного фрагмента в биологически активные структуры, уже применяемые лекарственные препараты (ингибиторы матричной металлопротеиназы, иммуносупрессоры, ингибиторы гистондеацетилазы и другие) значительно изменяет электронные свойства и повышает биологическую активность этих структур. [31-37]
4. Экспериментальная часть
Все использованные в работе растворители и реагенты являются коммерчески доступными. Используемые растворители очищали по стандартным методикам: MeCN перегоняли над P2O5, диоксан над Na. Абсолютный DMSO (хранящийся над молекулярными ситами 4А, Acros) использовали без дополнительной очистки. ГХ анализ проводили на хроматографе Хроматэк Кристалл-2000М на капиллярной колонке Macherey-Nagel OPTIMA-1 (30 м х 0.25 мм, поли(диметилсилоксан), 0.25 мкм), используя газ-носитель гелий и детектор ПИД. 1H и 13C ЯМР спектры регистрировались на приборах Bruker AC-200 (200.1 и 50.3 МГц, соответственно), Bruker AVANCE II 300 (300.1 и 75.4 МГц, соответственно) или Bruker AMX-400 (400.1 и 100.6 МГц, соответственно) в растворах в CDCl3, содержащих ТМС в качестве внутреннего стандарта, или в DMSO-d6. Химические сдвиги приведены в шкале м. д. в 1H спектрах относительно сигнала ТМС (? = 0.00 м. д.) или остаточного сигнала DMSO-d5 (? = 2.50 м. д.), а в 13C спектрах относительно сигнала CDCl3 (? = 77.1 м. д.) или DMSO-d6 (? =39.5 м. д.). 19F ЯМР спектры регистрировались на приборах Bruker AC-200 (188.3МГц) и Bruker AVANCE II 300 (282.4 МГц), химические сдвиги приведены относительно CFCl3 (? = 0.0 м. д.), используемого в качестве внешнего стандарта, или относительно 4-фторанизола (? = -125.0 м. д.), используемого в качестве внутреннего стандарта. 1H, 13C и 19F гомо - и гетероядерные корреляционные спектры регистрировались на приборе Bruker AVANCE II 300. Масс-спектры регистрировали на приборе Finnigan MAT INCOS-50 (ЭУ, 70 эВ). ГХ/МС анализ проводили на хроматографе Trace GC Ultra с FinniganMAT DSQ II масс-детектором (ЭУ, 70 эВ, 200°C).
4.1 Синтез силилзамещенных гем-фторбромциклопропанов
К смеси олефина, CHBr2F (2 экв.), ТЭБАХ (0.02 экв) и CH2Cl2 (0.50 мл на ммоль олефина) при перемешивании добавили 50% раствор NaOH (8.7 экв.). Реакционную смесь активно перемешивали на шейкере в течение 5-10 часов, контролируя конверсию методом ГХ. Реакционную смесь разбавили водой и CH2Cl2. Органический слой отделили, высушили над MgSO4. Растворитель отогнали на роторном испарителе, остаток перегнали в вакууме.
3-Фтор-3-бром-транс-1-фенил-2-(триметилсилил)циклопропан 1a
Из 0.8340 г (4.74 ммоль) ?-(триметилсилил)стирола, 1.8202 г (9.48 ммоль) CHBr2F, 0.0210 г (0.0925 ммоль) TЭБАХ, 2.36 мл CH2Cl2 и 1 мл (82.5 ммоль) 50% раствора NaOH в воде получили после перегонки 0.710 г 1a в виде желтого масла (син-1а/анти-1а =70/30, выход 52%, т. кип. 57-62°C/0,1 мм рт. ст., чистота по ЯМР и ГХ > 99%).
1H ЯМР (300.1 МГц, DMSO-d6) ?: (смесь син - и анти - изомеров) 0.16-0.21 (м. 9Н, Si(CH3)3), 1.42-1.64 (м., 1H, СHSiMe3-циклопроп.), 2.75-2.89 (м. 1Н, CH-циклопроп.), 7.22-7.40 (м., 5Н, аром.). 19F ЯМР (282.4 МГц, DMSO-d6): ?: -126.6 (д., 1F, син-1а, 3JHF(цис)=25.6 Гц), -119.0 (д. д., 1F, анти-1а, 3JHF(цис)=14.3 Гц, 3JHF(транс)=14.3 Гц).
2-Фтор-2-бром-1-фенил-1-(триметилсилил)циклопропан (1b)
Из 6.832 г (38.8 ммоль) ?-(триметилсилил)стирола, 12.50 г. (65.1 ммоль) CHBr2F, 0.1482 г (0.65 ммоль), 16.3 мл CH2Cl2 и 8.2 мл (338 ммоль) 50% раствора NaOH в воде получили 8.390 г 1b в виде желтого масла. (син-1b/анти-1b = 27/73, выход 75%, т. кип. 53-61°C/0,05 мм рт. ст., чистота по ЯМР и ГХ > 99%).
1Н ЯМР (300.1 МГц, DMSO-d6) ?: (смесь син - и анти - изомеров) 0.00 (д. 9Н, Si(CH3)3, анти-1b, 4JHF=1.0 Гц), 0.04 (с., 9Н, Si(CH3)3, син-1b), 1.68 (д. д., 1H, CH2-циклопроп.- анти-1b, 2JHH=7.1 Гц, 3JHF(транс) =4.4 Гц), 1.84 (д. д., 1Н, CH2-циклопроп., син-1b, 3JHF(транс)=8.3 Гц, 2JHH = 7.3 Гц), 1.92 (д. д., 1Н, CH2-циклопроп., син-1b, 3JHF(цис) = 16.8 Гц, 2JHH = 7.3 Гц), 2.13 (д. д., 1Н, CH2-циклопроп., анти-1b, 3JHF(цис) = 13.3 Гц, 2JHH=7.1 Гц), 7.14-7.38 (м. 5Н, оба изомера, аром.). 19F ЯМР (282.4 МГц, DMSO-d6): ?: -120.9 (уш. д. д. 1F, анти-1b, 3JHF(цис)=13.3 Гц, 3JHF(транс)=4.4 Гц), -119.5 (д. д., 1F, син-1b, 3JHF(цис)=16.8 Гц, 3JHF(транс)=8.4 Гц).
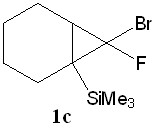
7-Фтор-7-бром-1-(триметилсилил)бицикло[4.1.0]гептан (1c)
Из 0.415 г (2.69 ммоль) 1-(триметилсилил)-циклогексена, 1.143 г (5.95 ммоль) CHBr2F, 4 мл CH2Cl2, 0.0129 г (0.057 ммоль) ТЭБАХ и 2 мл (52 ммоль) 50% раствора NaOH в воде получили 0.150 г (0.59 ммоль) вещества в виде желтого масла (син-1с/анти-1с = 23/77, выход 21%, чистота по ЯМР и ГХ>99%).
1Н ЯМР (200.1 МГц, CDCl3) ?: (смесь син - и анти-изомеров) 0.0–0.15 (м. 9Н, Si(CH3)3), 0.80-2.16 (м., 9H, CH-циклопроп., 4CH2), 19F ЯМР (188.3 МГц, CDCl3): ?: -132.7 (уш. с., 1F, син-1с), -112.5 (д., 1F, анти-1с, 3JHF(цис)= 16.2 Гц).
4.2 Циклопропанирование под действием CBr2FCO2Na
4.2.1. Синтез дибромфторацетата натрия
К раствору 20.6 г (198 ммоль) малоновой кислоты в 80 мл воды при перемешивании при комнатной температуре в течение 25 минут прикапывали 109.5 г (690 ммоль) Br2. Перемешивали при температуре 45–47°С с обратным холодильником в течение 44 часов. Упарили на роторном испарителе при 5–10 мм рт. ст. и т. бани 40–45°C, остаток воды отогнали с бензолом с насадкой Дина-Старка при т. бани 90–94°С. Полученное после упаривания растворителя желтое кристаллическое вещество растворили в 185 мл MeOH, при охлаждении на бане с холодной водой прикапали 14 мл (190 ммоль) SOCl2 за 75 минут. Реакционную смесь упарили на ротоном испарителе и перегнали при пониженном давлении, собирая фракцию с т. кип. 86-87°С/1 мм рт. ст. Получили 44.8 г CBr3CO2Me в виде бесцветной жидкости (выход 48%). К полученному эфиру добавили 8.8 г (49 ммоль) SbF3 и 5.7 г (35.6 ммоль) Br2. Реакционную смесь перемешивали при температуре 120°С с обратным холодильником и CaCl2-трубкой в течение 4 часов. Охладили до комнатной температуры, при перемешивании добавили петролейный эфир, H2O и Na2SO3 до обесцвечивания, органический слой экстрагировали и высушили над MgSO4. Растворитель отогнали при пониженном давлении, остаток перегнали, собирая фракцию с т. кип. 75-77°С / 45 мм рт. ст. Получили 14.3 г CBr2FCO2Me в виде бесцветной жидкости (выход 40%). К раствору 2.25 г (57 ммоль) NaOH в 61.5 мл EtOH при охлаждении на бане с холодной водой добавили полученный эфир. Реакционную смесь перемешивали при комнатной температуре. Растворитель упарили при пониженном давлении, остаток высушили в высоком вакууме (0.05 мм рт. ст., 20°С). Получили 11.21 г CBr2FCO2Na в виде белого кристаллического вещества (43.8 ммоль, выход 77%, общий выход на 4 стадии 22%). Содержание основного вещества по 19F ЯМР с внутренним стандартом (CF3CO2Na) >87%.
19F ЯМР (188.3 МГц, H2O) ?: -55.9 (с., 1F)
4.2.2. Оптимизация условий циклопропанирования
К раствору 0.0447 г (0.248 ммоль) 1,1-дифениэтилена в 1.0 мл диглима в атмосфере аргона при 100°C прикапывали раствор дибромфторацетата натрия 0.2616 г (1.01 ммоль) в 3.0 мл диглима в течение 10 минут. Перемешивали в течение 25 минут при 100°С. Реакционную смесь разбавили гексаном, промыли водой, высушили над MgSO4, упарили на роторном испарителе.
Определили конверсию по 1H ЯМР по отношению сигналов 1d (2.05-2.18 м. д., м. 2Н, CH2-циклопроп.) и 1,1-дифенилэтилена (5.21 м. д., с., 2Н, =CH2). Соотношение 1,1?дифенилэтилен/1d = 51/49.
Аналогично проводили реакции при разных температурах и соотношениях 1,1?дифенилэтилена и CBr2FCO2Na. Результаты приведены в таблице 2
4.2.3. Циклопропанирование (1-фенилэтенил)пинаколбороната
К раствору 23.2 мг (0.1 ммоль) (1-фенилэтенил)пинаколбороната в 0.5 мл диглима при перемешивании при температуре бани 100оС в течение 10 минут добавляли 102.8 г (0.4 ммоль, 4 экв.) твердого дибромфторацетата натрия. Перемешивали 5 минут, охладили до комнатной температуры.
2-Фтор-2-бром-1-фенил-1-[(пинакол)боронил]циклопропан (1e)
Выход 1e по ЯМР 29%
19F ЯМР (188.3 МГц, диглим) ?:-123.6(д. д.,1F, один изомер, 3JHF(цис)=10.0 Гц, 3JHF(транс)=5.3Гц), -129.7(м., 1F, другой изомер).
4.3 Изомеризация силилзамещенных гем-фторбромциклопропанов
2-фтор-3-бром-3-триметилсилил-1-фенилпропен (2a)
В ЯМР-ную ампулу поместили 0.0177 г (0.12 ммоль) бромида меди и 0.1441 г (0.502 ммоль) гем-фторбромциклопропана. В качестве внутреннего стандарта был добавлен 4-фторанизол. В атмосфере аргона прилили 0.50 мл MeCN, грели в течение 5 часов при 100°С. Выход по 19F ЯМР 87% (Z-2а/E-2a = 97/3).
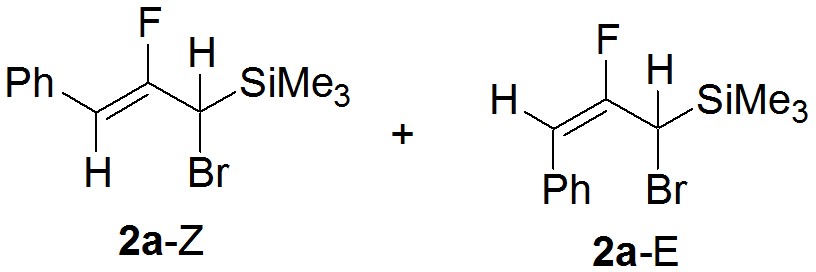
19F ЯМР (282.4 МГц, CH3CN): ?: -104.3(д. д., Z-2а, 3JHF(транс)= 38.1 Гц, 3JHF= 38.1 Гц), -101.2 (д. д. 1F, E-2а, JHF= 44.3 Гц, JHF= 19.4 Гц). 29Si ЯМР (59.6 МГц, CH3CN) ?: -7.25 (с., Z-2a), 7.48 (с., E-2a, из {1H,29Si}-HMBC)
2-фтор-3-бром-1-триметилсилил-1-фенилпропен (2b)
В ЯМР-ную ампулу поместили 0.0145 г (0.1 ммоль) бромида меди и 0.1394 г (0.49 ммоль) гем-фторбромциклопропана. В качестве внутреннего стандарта был добавлен 4-фторанизол. В атмосфере аргона прилили 0.5 мл MeCN, перемешивали в течение 48 часов при 100°С. Разбавили CH2Cl2, промывали водой, органический слой просушили над MgSO4, упарили на роторном испарителе при 5-10 мм рт. ст. (Выход по ЯМР 64%. Z/E=10/1).
19F ЯМР (282.4 МГц, CD3CN): ?: -92.9 (т., Z-3а, 3JHF= 21.3 Гц), -81.6 (т., E-3a, 3JHF= 22.5 Гц). 29Si ЯМР (59.6 МГц, CH3CN) ?: от -3.8 до -3.1 (м., оба изомера).
Для выделения 3a провели синтез в аналогичных условиях в отсутствии 4-фторанизола. Отделяли от примесей методом колоночной хроматографии (элюент н?гексан/бензол от 25/1 до 5/1). После КХ выделили всего 15 мг вещества, потому что большая часть разложилась на силикагеле.
5. Выводы
1. Осуществлен синтез неописанных ранее триметилсилилзамещенных гем-фторбромциклопропанов карбенным циклопропанированием ? - и ?-(триметилсилил)стиролов и 1?(триметилсилил)циклогексена в условиях щелочного дегидрогалогенирования дибромфторметана
2. Предложен новый реагент для фторбромциклопропанирования в нейтральных условиях — CBr2FCO2Na. С использованием 1,1-дифенилэтилена в качестве модельного субстрата проведена оптимизация условий проведения процесса. Показана возможность использования этого метода для циклопропанирования алкенил(пинакол)боронатов.
3. Изучены превращения 1-фенил-1-триметилсилил - (1a) и 1-фенил-2-триметилсилил-гем-фторбромциклопропанов (1b) с раскрытием цикла катализируемые CuBr в различных растворителях.
6. Литература
[1] W. K. Hagmann, J. Med. Chem. 51 (2008) 4359–4369.
[2] I. Ojima, Fluorine in Medicinal Chemistry and Chemical Biology, Chichester, UK, 2009, 640 p.
[3] E. P. Gillis, K. J. Eastman, M. D. Hill, D. J. Donnelly, N. A. Meanwell, J. Med. Chem. 58 (2015) 8315–8359.
[4] C. Isanbor, D. O’Hagan, Journal of Fluorine Chemistry 127 (2006) 303–319
[5] P. Shah, A. D. Westwell, Journal of Enzyme Inhibition and Medicinal Chemistry, 22 (2007) 527–540
[6] S. E. Thomas, Organic synthesis. The role of boron and silicon, Oxford University Press, New York, 1991, 97 p.
[7] T. K. Sarkar, Product subclass 40: Allylsilanes, Sci. Synth. 4 (2002) 837–925.
[8] A. Hosomi, K. Miura, Allylsilanes and related compounds, in: Compr. Organomet. Chem., Elsevier Ltd., 2008
[9] D. Seyferth, R. M. Simon, D. J. Sepelak, H. A. Klein, J. Org. Chem. 45 (1980) 2273–2274.
[10] D. Seyferth; K. R Wursthorn.; J. Organomet. Chem., 182 (1979) 455 – 464
[11] , Катализируемые соединениями меди превращения гем-фторхлор - и гем-фторбромциклопропанов с раскрытием цикла, диссертация на соискание ученой степени канд. хим. наук, Москва, 2016, 215 с.
[12] F. Tellier; J.-M. Duffault, M. Baudry, R. Sauvetre, J. Fluorine Chem.; 91 (1998) 133 – 139
[13] T. Hiyama, M. Obayashi, M. Sawahata, Tetrahedron Lett. 24 (1983) 4113–4116.
[14] Y. Xu, F. Jin, W. Huang, J. Org. Chem., 59 (1994) 2638 – 2641
[15] T. Hayashi, Y. Usuki, H. Iio, J. Fluorine Chem. 131 (2010) 709–713.
[16] R. Corbern, N. W. Mszar, A. H. Hoveyda, Angew. Chem. 123 (2011) 7217 –7220
[17] J. Lee, M. Tsukazaki, V. Snieckus, Tetrahedron Lett., 34 (1993) 415–418
[18] P. V. Ramachandran, A. Tafelska-Kaczmarek, A. Chatterjee, J. Org. Chem. 77 (2012) 9329?9333
[19] P. Martinet, R. Sauvetre, J.-F. Normant, J. Organomet. Chem., 367 (1989) 1–10
[20] P. Tarrant, W. H. Oliver, J. Org. Chem., 31 (1966) 1143–1146
[21] F. Tellier, R. Sauvetre, J.-F. Normant, J. Organomet. Chem., 362 (1989) 23–29
[22] C. Aouf, M. Santelli, Tetrahedron Lett., 52 (2011) 688 – 691
[23] O. M. Nefedov, N. V. Volchkov, Mendeleev Commun., 16 (2006) 121–128.
[24] M. Schlosser, Tetrahedron, 34 (1978) 3–17.
[25] , , Изв. АН СССР, Сер. хим. 27 (1978) 1168–1174.
[26] , , Ж. Орг. Хим., 29 (1993) 510–517
[27] N. V. Volchkov, M. A. Novikov, M. B. Lipkind, O. M. Nefedov, Mendeleev Commun. 23 (2013) 19–21.
[28] , , Изв. ак. наук, Сер. хим. 62 (2013) 71–82.
[29] M. A. Novikov, N. V. Volchkov, M. B. Lipkind, O. M. Nefedov, J. Fluorine Chem. 180 (2015) 131–143.
[30] Novikov M. A., Ibatov Y. A., Volchkov N. V., Lipkind M. B., Semenov S. E., Nefedov O. M., J. Fluor. Chem., 194 (2017) 58–72.
[31] S. L. Grage, X. Xu, M. Schmitt, P. Wadhwani, A. S. Ulrich J. Phys. Chem. Lett. 5 (2014) 4256?4259
[32] K. W. Laue, S. Kroger, E. Wegelius, G. Haufe Eur. J. Org. Chem. 2000, 3737-3743
[33] M. Lubke, M. Jung, G. Haufe J. Fluorine Chem. 152 (2013) 144–156
[34] M. Marhold, A.S. Ulrich, Wittmann, S. Grimme, T. Takahashi, G. Haufe, J. Fluorine Chem., 128 (2007) 1306–1317
[35] S. S. Schilson, P. Keul, R. S. Shaikh, M. Schafers, B. Levkau G. Haufe Bioorg. Med. Chem. 23 (2015) 1011–1026
[36] D. M. Shendage, R. Frohlich, K. Bergander, and G. Haufe Eur. J. Org. Chem. 2005, 719–727
[37] Y. Usuki, Y. Wakamatsu, M. Yabu, and H. Iio Asian J. Org. Chem. 2014, 270 – 1272
