
Партнерка на США и Канаду по недвижимости, выплаты в крипто
- 30% recurring commission
- Выплаты в USDT
- Вывод каждую неделю
- Комиссия до 5 лет за каждого referral
ГБОУ ЛГК на Юго – Востоке
(Московский Химический Лицей 1303)
Синтез производных 5-фтор, 5-фенилтриптамина, потенциальных противоопухолевых препаратов.
Выполнила:
Руководитель: к. х.н.
Москва 2017
Введение.
Триптамин — это моноаминный алкалоид, производное индола. Химически схож с аминокислотой триптофаном, поэтому часто встречается в организмах растений и животных и играет важную роль в их жизнедеятельности. Также триптамин выполняет функцию нейромедиатора и нейротрансмитера в головном мозге млекопитающих.
Серотонин имеет строение 5-гидрокситриптамина и часто называется «гормоном хорошего настроения». Является одним из основных из нейромедиаторов, играет важную роль в процессах свёртывания крови, участвует в процессах аллергии и воспаления.
Эти примеры хорошо показывают, что триптамин и его производные обладают широким спектром биологической активности. В настоящее время производные триптамина используются в качестве препаратов для лечения ожирения, аллергических реакций, мигрени и различных видов опухолей. Например, суматриптан, используется, как лекарство для лечения мигрени и индопан, как антидепрессант.
В организме человека печенью выделяется такое вещество, как моноамин оксидаза. Это фермент, осуществляющий катаболизм как эндогенных (нейромедиаторов и гормонов), так и экзогенных (попадающих в организм с пищей, лекарствами и психоактивными веществами) моноаминов посредством их окислительного дезаминирования. Такое поведение фермента ограничивает применение многих аминов в фармацевтике. Однако выяснено, что разветвление углеродной цепи в аминах способствует замедлению их метаболизма, что продлевает действие лекарственного препарата.
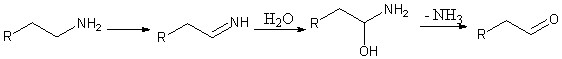
Целью нашего проэкта является синтез разветвленных производных 5-фтортриптамина, обладающих долгой препаративной активностью в человеческом организме.
Литературный обзор.
1. Synthesis of some 5-substituted-2-methyltryptamines and their N-mono - and-di-alkyl derivatives.
Авторы: Chapman N. В., Clarke К., Hughes H.
В статье Чапмана, Хьюза и Кларка, опубликованной в 1965 году, описывается метод синтеза 2-(1H-индол-3-ил-2-метил-5-фтор)-1-этан-1-амина. Вначале авторы используют перегруппировку по типу Фишера с участием катализатора хлорида цинка. К производному индола, полученному в данной реакции, добавляют оксалил хлорид. Реакция протекает с образованием 3-индолилглиоксилоил хлорида, после чего его конденсируют с помощью аммиака, метиламина, диметиламина или пирролидина. Полученныый амид восстанавливают гидридом алюминия-лития и получают замещенные триптамины, в том числе и 2-(1H-индол-3-ил-2-метил-5-фтор)-1-этан-1-амин.
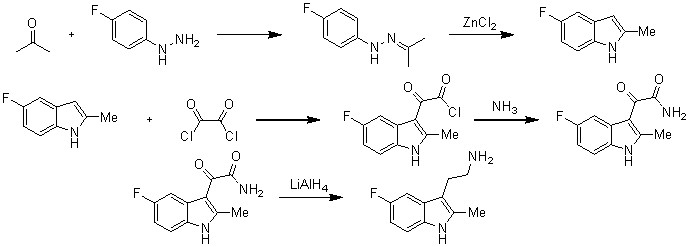
J. Chem. Soc., 1965, 1424
2. Синтез поизводных триптаминов и их циотоксичные свойства.
Авторы: Саликов Белый Хуснутдинова Вахитова Томилов
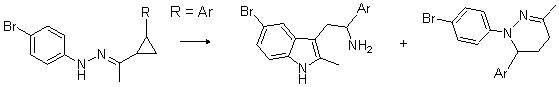
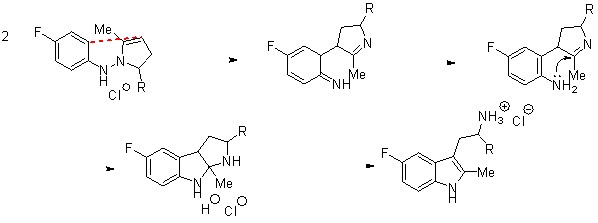
Здесь авторы применяют разработанную ими ранее перегруппировку гидразонов циклопропилкетонов для синтеза разветвленных триптаминов с использованием в качестве исходных циклопропилкетонов с заместителем во втором положении малого цикла. Использование в данной реакции 2-арилциклопропилэтанонов приводит к раскрытию малого цикла исключительно по наиболее замещенной связи с образованием б-арилтримптаминов в качестве основного продукта, а также небольшого количества замещенных тетрагидропиридазинов (схема 1).
Схема 1.
А также синтезируют множество производных 5-бромтриптамина путем замещения водородов, находящихся в различных положениях в соединении, на другие функциональные группы (схема 2) и 5-фенилтриптамин.
Схема 2.
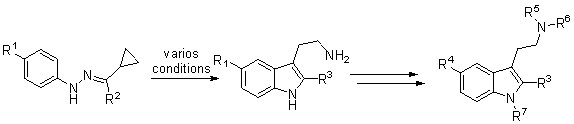
Схема 3.
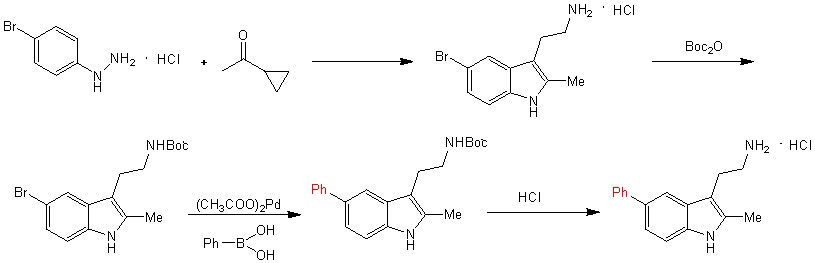
После получения большого числа производных 5-бромтриптамина, авторы исследуют их на различных клеточных линиях вследствие проявления антиопухолевой активности (таблица 1).
Таблица 1.
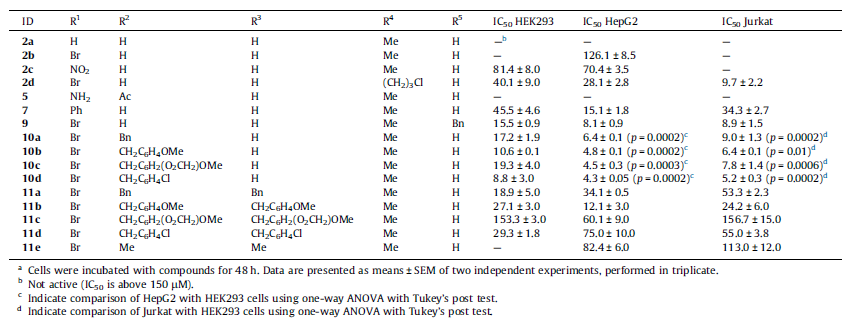
Bioorg. Med. Chem. Lett. 2015. 3597.
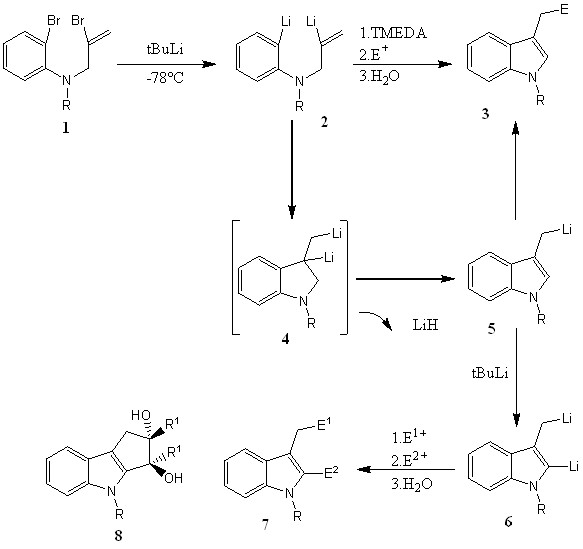
3. Synthesis of Functionalized Pyrrole and Indole Derivatives through
Carbometallation of Lithiated Double Bonds.
Авторы: Francisco J. FanД anaВs, Alejandro Granados, Roberto Sanz, JoseВ M. Ignacio, and JoseВ Barluenga.
Внутримолекулярное карбометаллирование 2-литий - N-(2-литийаллил)-аминов.
Замечания к процессам, изображенным ниже:
Добавление различных электрофилов в реакции 2-3 приводит к изоляции функциональных индолов;
Образование индольного центра (2-3) объясняется начальным карбометалированием виниллитиевого фрагмента ариллитием в дианионе 2;
Реакция с интермедиатом 5 с добавлением электрофила приводит к продукту 3;
С добавлением TMEDA (2-3), скорость реакции увеличиватся;
Внутримолекулярное карбометаллирование литированной двойной связи виниллитием проходит быстрее, чем ариллитием(9,10,11);
Однозащищенные индолы могут быть литированны по C2 с участием сильных оснований, таких как трет-бутиллитий;
Взаимодействие реагента 6 с 1,2-дикетонами дает селективный выход производных циклопенто-[b]индола (8) в виде единственного диостереоизомера;
Вышеописанный новый способ синтеза индолов с образованием связи C3 ± C3a имеет преимущество над циклизацией охало-N-аллила или N-виниланилина, катализируемые Pd, так как дальнейшая функционализация может быть проведена в ту же самую стадию реакции.
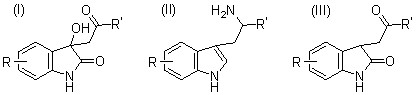
4. A Novel Preparation of alpha-Substituted Tryptamines from Isatins.
Авторы: C. S. Franklin, A. C. White.
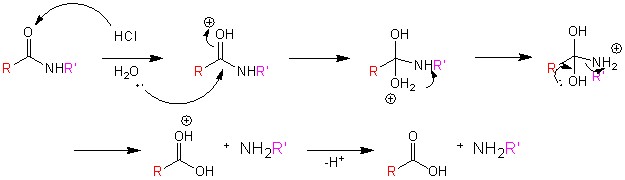
В этой статье авторы пишут, что диоксоиндолы (I) уже были получены конденсацией, катализируемой основанием, из изатинов и кетонов (Elderfield, "Hetrocyclic Compounds," New York, 1952, Vol. III, p. 222.) Восстановление их оксимов гидридом алюминия-лития привело к получению необходимых триптаминов (II), возникших в результате спонтанной дегидратации интермедиата, гидроксииндола. Также были синтезированны б - замещенные триптамины восстановлением оксимов из оксоиндолов (III) с добавлением этанола и натрия (Pietra and Tacconi, Farmaco (Pavia), 1958, 13, 893; 1961, 16 483, 492). В реакциях восстановления, проведенных во время подготовки данной научной работы, также был испозован комплекс хлорида алюминия и борогидрида натрия, который был наиболее эффективен в реакции с трифторометильным аналогом (II; R = 7-CF3, R' = Me). Он проявил себя, восстанавливая CF3– группу до Me–. Восстановление оксимов до аминов с использованием хлорида алюминия – борогидрида натрияранее не описывалось в литературе.
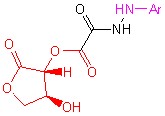
![]()
Обзор достигнутых результатов.
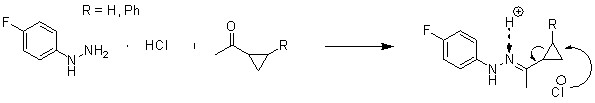
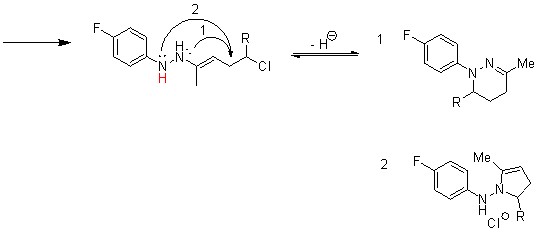
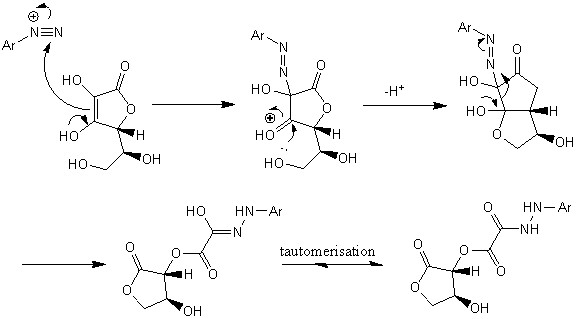
В данном проекте были синтезированы 2 производных 5-фтортриптамина с использованием ранее разработанной в нашей лаборатории (см. литературный обзор, №2) перегруппировки гидразонов циклопропилкетонов с использованием в качестве исходных циклопропилкетонов с заместителем во втором положении малого цикла (схема 1).
Схема 1.
Полученные производные были модифицированы с использованием восстановительного аминирования с образованием монобензилированных производных с алифатическим атомом азота через промежуточный продукт, являющийся имином (схема 2).
Схема 2.
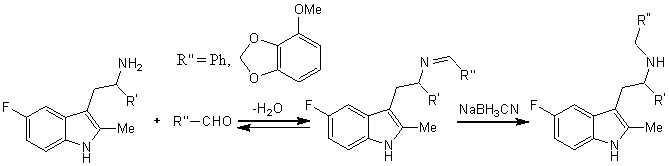
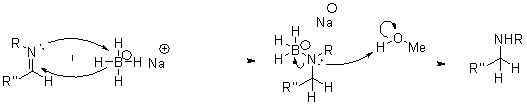
Также были проведены реакции (схема 3) для ‘защиты’ алифатических атомов водорода аминогруппы во избежание их замещения на функциональные единицы, которые будут введены в реакционную массу в последующих реакциях.
Схема 3.
R’ = H, Ph.
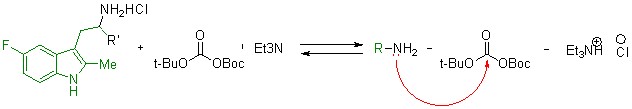
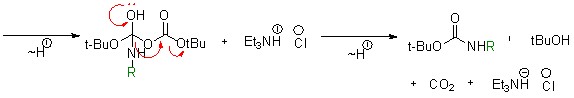
Полученные производные 5-фтортриптамина были отправлены на испытания противоопухолевой активности, так как ранее было обнаружено, что его 5-бромпроизводные обладают таковой.
В результате исследований, были получены следующие данные (таблица 1). Общая схема соединений для таблицы приведена ниже.
Таблица 1.
Вычисленные значения IC50, характеризующие параметры цитотоксичности (концентрация соединения, необходимая для 50 % ингибирования жизнеспособности клеток).
№ | R1 | R2 | R3 | IC50 HEK293, мкМ | IC50 HepG2, мкМ | IC50 Jurkat, мкМ |
1 | F | Ph | H | 73,46 | * b | * |
2 | F | Ph | CH2C6H2(O2CH2)OMe | 54,93 | * | * |
3 | F | H | CH2C6H2(O2CH2)OMe | 17,66 | * | * |
4 | H | Ph | H | 109,00 | * | * |
5 | Me | Ph | H | 94,04 | * | * |
6 | MeO | Ph | H | 80,29 | * | * |
7 | Br | H | H | - a | 126,1 ± 8,5 | - |
8 | Br | Ph | H | 39,37 ± 1,83 | 13,98 ± 1,92 | 20,41 ± 1,99 |
9 | Br | H | Bn | 17,2 ± 1,9 | 6,4 ± 0,1 | 9,0 ± 1,3 |
10 | Br | H | CH2C6H2(O2CH2)OMe | 19,3 ± 4,0 | 4,5 ± 0,3 | 7,8 ± 1,4 |
a Не активно (IC50 превышает 150 мкМ).
b Нет данных на сегодняшний день.
Описание таблицы:
- HEK293, HepG2, Jurkat – клеточные линии, на которых проводились испытания полученных веществ; HEK293 – клеточная линия, состоящая из условно здоровых клеток человека,
HepG2 и Jurkat состоят из опухолевых;
- мкМ – микромоль/л. Соединения 4-6 были синтезированы дополнительно по схеме 1. В качестве исходного реагента использовалось производное гидразина с соответствующими заместителями.
Чтобы синтезировать триптамины с заместителем фенилом в 5-ом положении, необходимо использовать методику, приведенную в статье №2, литературный обзор. Однако, этот метод недостаточно эффективен, так как на каждой стадии осуществляется потеря основного продукта, триптамина. Поэтому, мы решили ситезировать [1,1-бифенил]-4-гидразин, исходный компонент для реакции, изображенной на схеме 1, в несколько стадий (схема 4).
Схема 4.
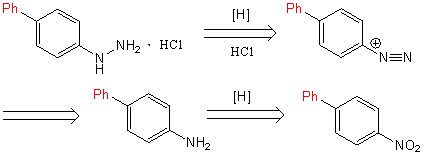
Осуществление превращения 4-нитро-1-бифенила в [1,1-бифенил]-4-амин происходило посредством гидрирования (схема 5).
Схема 5.
![]()
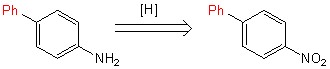
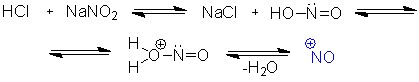
Переход [1,1-бифенил]-4-амина в [1,1-бифенил]-4-диазоний происходил с использованием частицы NO+, созданной в результате взаимодействия азотной и соляной кислот (схема 6).
Схема 6.
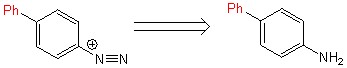
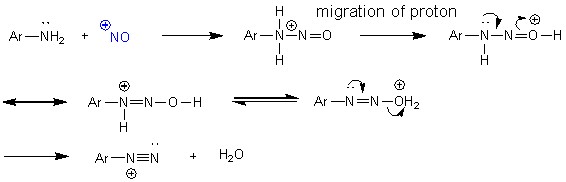
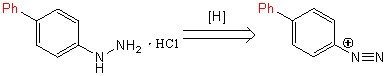
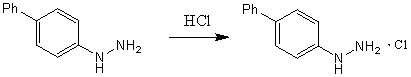
Восстановление [1,1-бифенил]-4-диазония до [1,1-бифенил]-4-гидразина происходило с участием аскорбиновой кислоты (ранее не описывалось в литературе) просредством ее присоединения-отщепления. Позже с [1,1-бифенил]-4-гидразином была проведена реакция гидрохлорирования с образованием гидрохлорида [1,1-бифенил]-4-гидразина (схема 7).
Схема 7.
Экспериментальная часть.
Синтез производных 5-фтортриптамина.
2-(1H-индол-3-ил-2-метил-5-фтор)-1-этил-1-амина гидрохлорид
К раствору 1 г (6.154 ммоль) гидрохлорида 4-фторфенилгидразина в 20 мл ацетонитрила добавили 0.62 г (7.385 ммоль) метилциклопропилкетона и кипятили реакционную массу с прямым холодильником в течение 16 часов при постоянном помешивании. После этого осадок отфильтровали от маточного раствора с помощью фильтра Шотта и сняли ЯМР 1Н спектр. В осадке наряду с продуктом обнаружилась примесь исходного гидрохлорида 4-фторфенилгидразина. Для того чтобы удалить примесь исходного вещества, осадок вернули в маточный раствор, добавили 0.31 г (3.691 ммоль) метилциклопропилкетона и продолжали кипятить реакционную смесь при постоянном помещивании в течение 10 часов, используя прямой холодильник.
По истечении этого времени, раствор профильтровали с помощью фильтра Шотта. Осадок просушили на вакууме. Получили 1.01 г гидрохлорида 2-(2-метил-5-фториндол-3-ил)-1-этиламина, выход 72%. Спектр ЯМР 1Н (200 MHz, ).08 (s), 8.16 (s), 7.24 (dd, J = 11.2, 7.3 Hz), 6.82 (dd, J = 13.3, 5.0 Hz), 3.49 (s), 2.93 (s), 2.42 (d, J = 33.3 Hz).
2-(1H-индол-3-ил-2-метил-5-фтор)-1-фенилэтил-1-амина гидрохлорид
К раствору 1 г (6.154 ммоль) гидрохлорида 4-фторфенилгидразина в 20 мл ацетонитрила добавили 1.18 г (7.385 ммоль) 2-фенилциклопропилэтанона и кипятили реакционную массу с обратным холодильником в течение 16 часов при постоянном помешивании. Раствор профильтровали через фильтр Шотта, оделив осадок, и сняли ЯМР 1Н спектр, тем самым обнаружив примесь иона аммония. Провели очистку продукта с помощью хроматографии (хлороформ/этиловый спирт = 9/1), просушили вещество на вакууме. Получили 0.72 г гидрохлорида 2-(2-метил-5-фториндол-3-ил)-1-фенилэтиламина, выход 38,5%. Спектр ЯМР 1Н (200 MHz, ).87 (s), 7.31 (s), 7.18 (d, J = 8.9 Hz), 6.92 – 6.53 (m), 4.26 (d, J = 5.4 Hz), 3.50 (s), 3.52 – 3.22 (m), 3.08 (d, J = 10.0 Hz), 2.51 (s), 1.84 (s), 1.06 (t, J = 7.0 Hz).
2-(1H-индол-3-ил-2-метил-5-фтор)-N-(метоксибензодиоксолил)метил-1-фенилэтил-1-амин
К раствору 0.20 г (0.657 ммоль) гидрохлорида 2-(1H-индол-3-ил-2-метил-5-фтор)-1-фенилэтил-1-амина в 5 мл ТГФ и 5 мл метилового спирта добавили 0.13 г (0.723 ммоль) метоксибензодиоксолилметил-5-карбальдегида, 0.08 г (1.314 ммоль) цианоборогидрида натрия, 0.12 г (1.970 ммоль) уксусной кислоты и 0.05 г (0.657 ммоль) ацетата натрия. Реакционную массу перемешивали в течение 24 часов, по истечении которых ее упарили. Сухой остаток растворили в хлороформе, полученный раствор перелили в делительную воронку. Туда же налили насыщенный раствор карбоната калия, закрыли воронку пробкой и тщательно встряхнули. После встряхивания, более плотную (нижнюю часть) разделившейся смеси экстрагировали, перелив в отдельную колбу. В оставшуюся в делительной воронке жидкость налили немного хлороформа и экстрагировали более плотную часть в колбу с продуктом реакции. Повторили эту операцию еще 2 раза, после чего вылили загрязненный раствор карбоната калия в слив. В делительную воронку налили раствор с продуктом реакции, добавив туда насыщенный раствор хлорида натрия, и проделали те же процедуры, что описаны выше. В колбу с продуктом добавили осушитель, сульфат натрия, и оставили на 20 минут, после профильтровав и упарив раствор. С вещества, оставшегося после упаривания, сняли ЯМР 1Н спектр, обнаружив при этом примеси помимо основного продукта. Чтобы избавиться от побочных продуктов, провели очистку вещества с помощью хроматографии (хлороформ/этиловый спирт = 9/1) и, упарив раствор, получили 0.25 г 2-(1H-индол-3-ил-2-метил-5-фтор)-N-(метоксибензодиоксолил)метил-1-фенилэтил-1-амина, выход 88%. Спектр ЯМР 1Н (200 MHz, ).72 (s), 7.29 (d, J = 12.0 Hz), 7.14 (s), 6.85 (s), 6.30 (s), 5.94 (d, J = 11.4 Hz), 3.92 (d, J = 6.3 Hz), 3.77 (s), 3.37 (d, J = 13.5 Hz), 2.94 (d, J = 5.3 Hz), 2.10 (s).
2-(1H-индол-3-ил-2-метил-5-фтор)-N-(метоксибензодиоксолил)метил-1-фенилэтил-1-амина гидрохлорид
К раствору 0.25 г (0.579 ммоль) 2-(1H-индол-3-ил-2-метил-5-фтор)-N-(метоксибензодиоксолил)метил-1-фенилэтил-1-амина в 20 мл этилового эфира добавили 0.723 мл 4М раствора соляной кислоты в диоксане. Реакционную смесь оставили покоиться до выпадения осадка. Осадок отделили от остального раствора с помощью фильтра Шотта и получили 0.20 г гидрохлорида 2-(1H-индол-3-ил-2-метил-5-фтор)-N-(метоксибензодиоксолил)метил-1-фенилэтил-1-амина, выход 75%.
2-(1H-индол-3-ил-2-метил-5-фтор)-N-(метоксибензодиоксолил)метил-1-этил-1-амин
К раствору 0.20 г (0.875 ммоль) гидрохлорида 2-(1H-индол-3-ил-2-метил-5-фтор)-1-этил-1-амина в 5 мл ТГФ и 5 мл метилового спирта добавили 0.17 г (0.963 ммоль) метоксибензодиоксолилметил-5-карбальдегида, 0.11 г (1.750 ммоль) цианоборогидрида натрия и 0.16 г (2.626 ммоль) уксусной кислоты. Реакционную смесь перемешивали в течение 24 часов, по истечении которых ее упарили. Сухой остаток растворили в хлороформе, полученный раствор перелили в делительную воронку. Туда же налили насыщенный раствор карбоната калия, закрыли воронку пробкой и тщательно встряхнули. После встряхивания, более плотную (нижнюю часть) разделившейся смеси экстрагировали, перелив в отдельную колбу. В оставшуюся в делительной воронке жидкость налили немного хлороформа и экстрагировали более плотную часть в колбу с продуктом реакции. Повторили эту операцию еще 2 раза, после чего вылили загрязненный раствор карбоната калия в слив. В делительную воронку налили раствор с продуктом реакции, добавив туда насыщенный раствор хлорида натрия, и проделали те же процедуры, что описаны выше. В колбу с продуктом добавили осушитель, сульфат натрия, и оставили на 20 минут, после профильтровав и упарив раствор. С вещества, оставшегося после упаривания, сняли ЯМР 1Н спектр, обнаружив при этом примеси помимо основного продукта. Чтобы избавиться от побочных продуктов, провели очистку вещества с помощью хроматографии (хлороформ/этиловый спирт = 9/1) и, упарив раствор, получили 0.27 г 2-(1H-индол-3-ил-2-метил-5-фтор)-N-(метоксибензодиоксолил)метил-1-этил-1-амина, выход 87%. Спектр ЯМР 1Н (200 MHz, ).78 (s), 7.26 (s), 7.23 – 7.05 (m), 6.85 (t, J = 9.0 Hz), 6.46 (s), 5.93 (s), 3.85 (s), 2.88 (s), 2.39 (s).
2-(1H-индол-3-ил-2-метил-5-фтор)-N-(метоксибензодиоксолил)метил-1-этил-1-амина гидрохлорид
К раствору 0.27 г (0.758 ммоль) 2-(1H-индол-3-ил-2-метил-5-фтор)-N-(метоксибензодиоксолил)метил-1-этил-1-амина в 20 мл ТГФ добавили 0.758 мл 4М раствора соляной кислоты в диоксане. Реакционную смесь оставили покоиться до выпадения осадка. Осадок отделили от остального раствора с помощью фильтра Шотта и получили 0.22 г гидрохлорида 2-(1H-индол-3-ил-2-метил-5-фтор)-N-(метоксибензодиоксолил)метил-1-этил-1-амина, выход 74%.
2-(((2-(1H-индол-3-ил-2-метил-5-фтор)этил)амино)метил)фенол
К раствору 0.20 г (0.875 ммоль) гидрохлорида 2-(1H-индол-3-ил-2-метил-5-фтор)-1-этил-1-амина в 10 мл метилового спирта добавили 0.12 г (0.963 ммоль) салицилового альдегида, 0.11 г (1.750 ммоль) цианоборогидрида натрия и 0.16 г (2.626 ммоль) уксусной кислоты. Реакционную смесь перемешивали в течение 24 часов, по истечении которых ее упарили. Сухой остаток растворили в хлороформе, полученный раствор перелили в делительную воронку. Туда же налили насыщенный раствор гидрокарбоната натрия, закрыли воронку пробкой и тщательно встряхнули. После встряхивания, более плотную (нижнюю часть) разделившейся смеси экстрагировали, перелив в отдельную колбу. В оставшуюся в делительной воронке жидкость налили немного хлороформа и экстрагировали более плотную часть в колбу с продуктом реакции. Повторили эту операцию еще 2 раза, после чего вылили загрязненный раствор гидрокарбоната натрия в слив. В делительную воронку налили раствор с продуктом реакции, добавив туда насыщенный раствор хлорида натрия, и проделали те же процедуры, что описаны выше. В колбу с продуктом добавили осушитель, сульфат натрия, и оставили на 20 минут, после профильтровав и упарив раствор. С вещества, оставшегося после упаривания, сняли ЯМР 1Н спектр, обнаружив при этом примеси помимо основного продукта. Чтобы избавиться от побочных продуктов, провели очистку вещества с помощью хроматографии (хлороформ/этиловый спирт = 9/1) и, упарив раствор, получили 0.04 г 2-(((2-(1H-индол-3-ил-2-метил-5-фтор)этил)амино)метил)фенола, выход 15%.
N-бензил-2-(1H-индол-3-ил-2-метил-5-фтор)-1-фенилэтил-1-амин
К раствору 0.20 г (0.657 ммоль) гидрохлорида 2-(1H-индол-3-ил-2-метил-5-фтор)-1-фенилэтил-1-амина в 10 мл метилового спирта добавили 0.15 г (1.445 ммоль) бензальдегида, 0.12 г (1.970 ммоль) уксусной кислоты, 0.05 г (0.657 ммоль) ацетата натрия и 0.08 г (1.313 ммоль) цианоборогидрида натрия. Реакционную массу перемещивали в течение 24 часов, после чего сняли ЯМР 1Н спектр. Обнаружив примеси, провели очистку вщества с помощью хроматографии (хлороформ/этиловый спирт = 14/1) и упарили раствор. Так как разделение вещества прошло не полностью, провели повторную очистку вщества с помощью хроматографии (хлороформ/этиловый спирт = 14/1), после чего провели третичную чистку вещества по тем же причинам (хлороформ/этилацетат = 7/1). В настоящее время вещество все еще не полностью очищено.
N-бензил-2-(1H-индол-3-ил-2-метил-5-фтор)этил-1-амин
К раствору 0.30 г (1.313 ммоль) гидрохлорида 2-(1H-индол-3-ил-2-метил-5-фтор)-1-этил-1-амина в 10 мл метилового спирта добавили 0.14 г (1.444 ммоль) бензальдегида, 0.24 г (3.929 ммоль) уксусной кислоты, 0.17 г (2.626 ммоль) цианоборогидрида натрия и 0.10 г (1.313 ммоль) ацетата натрия. Реакционную смесь перемешивали в течение 24 часов, по истечении которых ее упарили. Сухой остаток растворили в хлороформе, полученный раствор перелили в делительную воронку. Туда же налили насыщенный раствор гидрокарбоната натрия, закрыли воронку пробкой и тщательно встряхнули. После встряхивания, более плотную (нижнюю часть) разделившейся смеси экстрагировали, перелив в отдельную колбу. В оставшуюся в делительной воронке жидкость налили немного хлороформа и экстрагировали более плотную часть в колбу с продуктом реакции. Повторили эту операцию еще 2 раза, после чего вылили загрязненный раствор гидрокарбоната натрия в слив. В делительную воронку налили раствор с продуктом реакции, добавив туда насыщенный раствор хлорида натрия, и проделали те же процедуры, что описаны выше. В колбу с продуктом добавили осушитель, сульфат натрия, и оставили на 20 минут, после профильтровав и упарив раствор. С вещества, оставшегося после упаривания, сняли ЯМР 1Н спектр, обнаружив при этом примеси помимо основного продукта. Чтобы избавиться от побочных продуктов, провели очистку вещества с помощью хроматографии (хлороформ/этиловый спирт = 4/1) и, упарив раствор, получили 0.23 г N-бензил-2-(1H-индол-3-ил-2-метил-5-фтор)этил-1-амина, выход 63%. ЯМР 1Н спектр (200 MHz, ).97 (s), 7.41 – 7.24 (m), 7.15 (s), 6.83 (td, J = 9.2, 2.5 Hz), 3.86 (s), 2.93 (s), 2.34 (s).
N-бензил-2-(1H-индол-3-ил-2-метил-5-фтор)этил-1-амина гидрохлорид
К раствору 0.26 г (0.928 ммоль) N-бензил-2-(1H-индол-3-ил-2-метил-5-фтор)этил-1-амина в 20 мл ТГФ добавили 1.16 мл 4М раствора соляной кислоты в диоксане. Реакционную смесь поставили в холодильник до выпадения осадка, после чего осадок отфильтровали с помощью фильтра Шотта, оставили сушиться на воздухе ~ на 24 часа и получили 0.20 г гидрохлорида N-бензил-2-(1H-индол-3-ил-2-метил-5-фтор)этил-1-амина, выход 62%.
Трет-бутил (2-(1H-индол-3-ил-2-метил-5-фтор)-1-этил)карбомид
К раствору 0.38 г (1.680 ммоль) гидрохлорида 2-(1H-индол-3-ил-2-метил-5-фтор)-1-этил-1-амина в 20 мл дихлорметана добавили 0.19 г (1.848 ммоль) трэтиламина, 0.4 г (1.848 ммоль) ди-трет-бутилдикарбоната. Реакционную массу помешивали в течение получаса, после чего в нее добавили еще 0.19 г (1.848 ммоль) триэтиламина. Суммарное время помешивания = 4 ч. По истечении 4 часов, реакционную массу упарили. Сухой остаток растворили в хлороформе, полученный раствор перелили в делительную воронку. Туда же налили насыщенный раствор гидрокарбоната натрия, закрыли воронку пробкой и тщательно встряхнули. После встряхивания, более плотную (нижнюю часть) разделившейся смеси экстрагировали, перелив в отдельную колбу. В оставшуюся в делительной воронке жидкость налили немного хлороформа и экстрагировали более плотную часть в колбу с продуктом реакции. Повторили эту операцию еще 2 раза, после чего вылили загрязненный раствор гидрокарбоната натрия в слив. В делительную воронку налили раствор с продуктом реакции, добавив туда насыщенный раствор хлорида натрия, и проделали те же процедуры, что описаны выше. В колбу с продуктом добавили осушитель, сульфат натрия, и оставили на 40 минут, после профильтровав и упарив раствор. С вещества, оставшегося после упаривания, сняли ЯМР 1Н спектр, обнаружив при этом примеси помимо основного продукта. Чтобы избавиться от побочных продуктов, провели очистку вещества с помощью хроматографии (хлороформ/этиловый спирт = 9/1) и, упарив раствор, получили 0.514 г вещества, с которого сняли ЯМР 1Н спектр, повторно обнаружив примеси. Попытка очистить вещество путем перекристаллизации в системе (хлороформ/гексан = 2/1) при температуре ~-20°C оказалась безуспешной. Продукт не выделен до конца.
Трет-бутил (2-(1H-индол-3-ил-2-метил-5-фтор)-1-фенилэтил)карбомид
К раствору 0.30 г (0.998 ммоль) гидрохлорида 2-(1H-индол-3-ил-2-метил-5-фтор)-1-фенилэтил-1-амина в 20 мл дихлорметана добавили 0.11 г (1.098 ммоль) трэтиламина, 0.24 г (1.098 ммоль) ди-трет-бутилдикарбоната. Реакционную массу помешивали в течение получаса, после чего в нее добавили еще 0.26 г (2.545 ммоль) триэтиламинаСуммарное время помешивания = 4 ч. По истечении 4 часов, реакционную массу упарили. Сухой остаток растворили в хлороформе, полученный раствор перелили в делительную воронку. Туда же налили насыщенный раствор гидрокарбоната натрия, закрыли воронку пробкой и тщательно встряхнули. После встряхивания, более плотную (нижнюю часть) разделившейся смеси экстрагировали, перелив в отдельную колбу. В оставшуюся в делительной воронке жидкость налили немного хлороформа и экстрагировали более плотную часть в колбу с продуктом реакции. Повторили эту операцию еще 2 раза, после чего вылили загрязненный раствор гидрокарбоната натрия в слив. В делительную воронку налили раствор с продуктом реакции, добавив туда насыщенный раствор хлорида натрия, и проделали те же процедуры, что описаны выше. В колбу с продуктом добавили осушитель, сульфат натрия, и оставили на 40 минут, после профильтровав и упарив раствор. С вещества, оставшегося после упаривания, сняли ЯМР 1Н спектр, обнаружив при этом примеси помимо основного продукта. Чтобы избавиться от побочных продуктов, провели очистку вещества с помощью хроматографии (хлороформ/этиловый спирт = 9/1) и, упарив раствор, получили 0.33 г вещества, с которого сняли ЯМР 1Н спектр, повторно обнаружив примеси. Попытка очистить вещество путем перекристаллизации в системе (хлороформ/гексан = 2/1) при температуре ~-20°C оказалась безуспешной. Провели очистку вещества с помощью хроматографии (хлороформ/этиловый спирт = 9/1) и, упарив раствор, получили 0.34 г Трет-бутил (2-(1H-индол-3-ил-2-метил-5-фтор)-1-фенилэтил)карбомида, выход 98%.
[1,1-бифенил]-4-амин
К раствору 9.60 г (48.236 ммоль) 4-нитро-1-бифенила в 100 мл этилового спирта добавили 6.03 г (0.121 моль) гидразингидрата и 1 г суспензии катализатора Никеля Ренея в этиловом спирте (Ni/EtOH~1:1). Реакционную массу помешивали до тех пор, пока из нее не прекратил выделяться азот. После выявления с помощью ТСХ, что реакция не прошла полностью, в реакционную смесь добавили 6 мл гидразингидрата и продолжили помешивать, периодически добавляя малы порции катализатора. После того как прекратил выделяться азот, реакционную массу поставили в холодильник. Через несколько часов в колбе образовались кристаллы. Кристаллы были растворены посредством нагревания, полученный раствор был профильтрован от гетерогенной фазы (катализатора). Фильтрат слегка упарили и поставили в холодильник приблизительно на 5 часов, по истечении которых из раствора выпал осадок. Осадок отфильтровали, отжали, перенесли в чашку Петри, оставили сушиться на воздухе в течение нескольких часов и получили 6.16 г [1,1-бифенил]-4-амина, выход 76%.
Выводы.
፠ В данном проекте был синтезирован 5-фтортиптамин, несколько его разветвленных производных, а также триптамины с заместителями в 5-ом положении: H, Me, MeO – дополнительно.
፠ Эти соединения были проверены на цитотоксичность относительно условно здоровых клеток. Наименее токсичные свойства проявил водород-замещенный триптамин, показав результат 109.00; наилучшим показателем среди фтор-замещенных триптаминов является результат 73.46; тогда как для бром-замещенных триптаминов наилучший результат – 39.37; что показывает большую эффективность исследуемых соединений по сравнению с ранее изученными.
፠ Для синтеза 5-фенилтиптамина и его производных, удобным способом был получен гидрохлорид [1,1-бифенил]-4-гидразина, поскольку ранее использовавщийся способ синтеза триптаминов с заместителем Ph в 5-ом положении не является целесообразным вследствие потери необходимого нам вещества на каждой стадии.
