
Партнерка на США и Канаду по недвижимости, выплаты в крипто
- 30% recurring commission
- Выплаты в USDT
- Вывод каждую неделю
- Комиссия до 5 лет за каждого referral
ХИМИЧЕСКАЯ СВЯЗЬ В КООРДИНАЦИОННЫХ
СОЕДИНЕНИЯХ. ТЕОРИЯ ВАЛЕНТНЫХ СВЯЗЕЙ
При описании химической связи используются различные теоретические модели, появившиеся в разное время. Некоторые из них считаются современными (квантово-механические модели), некоторые устаревшими, хотя ими до сих пор пользуются.
Одной из важнейших является ионно-ковалентная модель. Ее возникновение связывают с именами В. Косселя и Дж. Льюиса (1916 г.).
Современные представления об электронном строении координационных соединений основано на квантовой механике. Квантово-механические модели из-за своих высоких и до конца еще непонятных потенциальных возможностей в настоящее время настолько популярны, что представляются стержнем существующего знания. Кроме того, в настоящее время общепринятым считается отсутствие необходимости в какой-то специальной теории координационной связи, хотя гипотезы о некоей ее специфике в свое время возникали. Еще в 70-е годы вполне серьезно говорили о донорно-акцепторной связи, как о координационной. Но важно понять, что донорно-акцепторная – одна из полуэмпирических моделей, множество которых разрабатывалось на ранних этапах развития квантовой химии, когда вычислительные ресурсы были крайне ограничены и когда проще было придумать новый алгоритм вычислений, основанный на эмпирических параметрах, чем провести собственно расчет.
В то же время даже сейчас при наличии мощной компьютерной техники подобные модели продолжают использоваться, хотя все более популярным становится применение неэмпирических методик для решения разных задач. Необходимым для массовых (недорогих и быстрых) расчетов является адиабатическое приближение, не учитывающее вибронные взаимодействия (взаимодействие электронов и ядер в молекуле). Для массовых расчетов электронного строения молекул применяют одноэлектронное приближение, недостатком которого является недоучет межэлектронного взаимодействия. В этом приближении движение электронов рассматривается при фиксированных положениях ядер.
Примечание 1. Скорости движения з в легких атомах, а также молекулах, состоящих из легких атомов, и тяжелых атомах и соответствующих молекул существенно различаются. В тяжелых атомах скорости движения з становятся сопоставимыми со скоростью света, поэтому на свойства тяжелых атомов и молекул существенно влияют релятивистские эффекты (магнитное спин-орбитальное взаимодействие, увеличение массы тела, замедление времени). Появление «благородных» свойств у тяжелых переходных металлов (серебро, золото, полладий, платина и др.) полностью определяются релятивистскими эффектами.
Для молекул, состоящих из легких атомов, релятивистские эффекты дают обычно меньший вклад, чем точность, с которой эти свойства измеряются.
Примечание 2. Наиболее популярным (ab inifio) методом расчета на протяжении трех последних десятилетий является МО ЛКАО или более полно – метод самосогласованного поля Хартри – Фока – Рутаана, обозначаемый в ангоязычной литературе SCF HFR (Self Consisted Field and Hartre – Fock – Roothaan). Точность метода зависит от выбранного для расчетов атомного базиса, методов учета энергии корреляции и методов учета релятивистских эффектов. В квантово-химических расчетах энергии образования сравнительно небольших молекул, состоящих из атомов трех первых периодов, достигнута точность ≈ 1 ккал/моль.
Значительная трудность применения метода МО ЛКАО – это сильная зависимость времени расчета от размеров атомных базисов (приблизительно N4, N – количество атомных орбиталей). В такой пропорции возрастает количество вычисляемых интегралов и, соответственно, стоимость расчетов. Поэтому большие усилия направлены на разработку методов, в которых не было бы такой сильной зависимости объема расчетов от размеров молекул.
Наиболее перспективным в этом отношении является метод, основанный на теории функционала плотности (ТФП), обозначаемой в научной литератере аббревиатурой DFT (от англоязычного названия Density Functional Theory). Преимущество этого метода состоит в значительно меньшем росте объема вычислений с ростом размера молекул при сохранении или даже увеличении точности расчетов по сравнению с методом ССП ХФР. Сегодня реализованы алгоритмы расчетов с зависимостью N![]() и испытываются алгоритмы расчетов с зависимостью N
и испытываются алгоритмы расчетов с зависимостью N![]() , т. е. на два порядка сокращается объем вычислений по сравнению с методом ССП ХФР. Поэтому методом ТФП успешно рассчитывают свойства не только достаточно больших молекул, но и кристаллов, полимеров, поверхностных слоев адсорбированных веществ и т. д. Для расчетов свойств веществ в конденсированных фазах этот метод не имеет конкурентов.
, т. е. на два порядка сокращается объем вычислений по сравнению с методом ССП ХФР. Поэтому методом ТФП успешно рассчитывают свойства не только достаточно больших молекул, но и кристаллов, полимеров, поверхностных слоев адсорбированных веществ и т. д. Для расчетов свойств веществ в конденсированных фазах этот метод не имеет конкурентов.
Быстрый рост вычислительных возможностей компьютерной техники позволил эффективно заниматься дизайном соединений с требуемыми свойствами, инженерией кристаллов и других материалов. Квантово-химическое моделирование свойств соединений стало рабочим инструментом в большинстве химических лабораторий.
ЭЛЕКТРОННОЕ СТРОЕНИЕ КООРДИНАЦИОННЫХ СОЕДИНЕНИЙ. ОСНОВНЫЕ ПОНЯТИЯ
В зависимости от типа валентных орбиталей и атомной массы элементы делят на несколько групп.
В атомах легких элементов основными взаимодействиями, определяющими их химические свойства, являются электростатическое притяжение электронов к ядру и межэлектронное взаимодействие. В атомах тяжелых элементов к этим взаимодействиям добавляются релятивистские эффекты, в частности, магнитное спин-орбитальное взаимодействие. Следовательно, строение молекул, состоящих из тяжелых атомов, должно рассматриваться с учетом этого дополнительного взаимодействия. Разумеется, резкой границы между «легкими» и «тяжелыми» атомами провести невозможно. Качественно принято считать: элементы верхней трети Периодической системы «легкими». Для них релятивистские эффекты в первом приближении можно не учитывать. Свойства элементов нижней трети Периодической системы нельзя количественно описать без учета релятивистских эффектов. Средняя треть элементов Периодической системы наиболее трудна для качественного описания свойств. Разные свойства этих элементов и их соединений по-разному зависят от релятивистских эффектов.
По типу валентных орбиталей элементы делятся на непереходные s-, p-элементы (элементы подгрупп А, элементы главных подгрупп), переходные d-элементы (элементы подгрупп Б), лантаноиды и актиноиды (f-элементы).
Специфику строения переходных металлов и их комплексов определяет наличие валентных nd-орбиталей. Общую электронную конфигурацию атомов переходных металлов можно написать так: Ar nsa (n-1) db, где Ar – замкнутая оболочка аргона, a = 0, 1, 2; b = 1, 2, …, 10. Квантово-химические расчеты показали, что (n – 1) d-орбитали плотнее окружают ядро и в среднем расположены ближе к ядру, чем ns-орбитали, что проиллюстрировано на рисунке (f – квантовое число).
В соответствии с меньшим средним расстоянием (n – 1) d-орбиталей от ядра они более стабильны, чем ns-орбитали, что называют орбитальным эффектом. Именно такой порядок уровней получается при расчетах методом Хартри – Фока – Рутаана, в которых игнорируется энергия корреляции электронов. Однако на энергию состояний атомов, соответствующих определенным конфигурациям: ns2 (n – 1) dq, ns1 (n – 1) d(q+1), ns0 (n – 1) d(q+2), влияет энергия корреляции. Поскольку ns-орбитали более диффузны, межэлектронное взаимодействие в состоянии ns2 меньше, чем в состоянии (n–1)dq. Таким образом, конкуренция двух противоположных по влиянию на энергию уровней фактора – орбитального эффекта и энергии межэлектронного взаимодействия, определяет энергетическую выгодность тех или других конфигураций переходных металлов.
Электронные конфигурации переходных металлов IV, V и VI периодов в основном состоянии приведены ниже.
IV период (3dn4sm)
Sc | Ti | V | Cr | Mn | Fe | Co | Ni | Cu | Zn |
d1s2 | d2s2 | d3s2 | d5s1 | d5s2 | d6s2 | d7s2 | d8s2 | d10s1 | 3d104s2 |
V период (4dn5sm)
Y | Zn | Nb | Mo | Tc | Ru | Rh | Pd | Ag | Cd |
d1s2 | d2s2 | d3s2 | d5s1 | d5s2 | d7s1 | d8s1 | d10 | d10s1 | 4d105s2 |
VI период (5dn6sm)
La | Hf | Ta | W | Re | Os | Ir | Pt | Au | Hg |
d1s2 | d2s2 | d3s2 | d4s2 | d5s2 | d6s2 | d7s2 | d9s1 | d10s1 | d10s2 |
Из приведенных конфигураций видно, что в отличие от переходных металлов IV периода, где только дважды нарушается порядок заполнения d, s-орбиталей, в атомах Cr и Cu, в элементах V периода этот порядок нарушается 5 раз. Как видно из данных, приведенных на рисунках…, орбитальные радиусы и энергии уменьшаются с ростом заряда ядра. Отклонения от монотонной зависимости на этих кривых связаны с изменением электронной конфигурации основного состояния. Например, для Cr и Cu реализуются конфигурации 4s13dn, тогда как для других переходных металлов этого периода конфигурациями основного состояния является 4s23d(n-1).
Разность орбитальных энергий ns и (n – 1) d уменьшается с ростом номера периода. Увеличение радиусов (n – 1) d-орбиталей и сближение их энергий с энергиями ns-орбиталей с ростом номера периода благоприятст-вует росту их валентной активности, что проявляется, например, в повышении относительной устойчивости высших степеней окисления более тяжелых элементов – аналогов, например: V – Nb – Ta и Cr – Mo – W.
Для самых тяжелых переходных металлов VI периода заметной становится также зависимость орбитальных энергий от квантового числа j, что является проявлением релятивистского эффекта.
Особенности строения атомов и ионов переходных металлов хорошо иллюстрируют энергии возбуждения электронов с (n – 1) d - на ns- и nр-ор-битали. Данные, рассчитанные из экспериментальных спектров атомов, приведены на рис. … Энергии возбуждения нейтральных атомов приведены на рис. …, а и аналогичныве данные для однозарядных и двухзарядных ионов – соответственно на рис. …, б и …., в.
Обратим внимание на немонотонный ход энергий возбуждения 3d(n-1)4s2 → 3dn4s1 в атомах переходных металлов IV периода. Отклонение от монотонности наблюдается (как и для средних радиусов и орбитальных энергий) на атомах Cr и Cu. Разность орбитальных энергий 3d и 4s иногда настолько мала, что относительная энергия конфигураций зависит от суммарного влияния двух взаимодействий: притяжения электронов к ядру (орбитальная энергия) и межэлектронного взаимодействия, включая энергию корреляции, зависящую от суммарного спина.
Рассмотрим электронные конфигурации и схемы заполнения уровней атомов V, Cr, Cu:
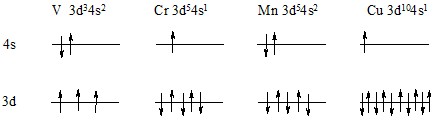
Упомянутая конкуренция двух видов взаимодействия (притяжение электронов к ядру и межэлектронное отталкивание) приводит к существованию частично заполненного уровня, расположенного ниже заполненного уровня. Это кажущееся противоречие связано со стремлением охарактеризовать строение сложных атомов в терминах водородоподобных (одноэлектронных) орбиталей и соответствующих им энергетических уровней. Обратим внимание на разность энергий атомов с конфигурациями 3d(n-1)4s2 и 3dn4s1 нейтральных атомов V и Ni (рис. …, а) или однозарядных катионов Ni, V, Fe и Co 3d(n-1)4s1 – 3dn (рис. …, б). Эти разности близки к нулю, и поэтому попытка охарактеризовать строение этих атомов переходных металлов электронными конфигурациями в терминах водородоподобных орбиталей и соответствующих уровней не имеет смысла.
Ионизация атомов переходных металлов в большей степени стабилизирует d-орбитали. В двухзарядных ионах конфигурация 3dn всегда стабильнее, чем конфигурация 3d(n-1)4s1 (рис. …, в).
Таким образом, используя электронные конфигурации атомов переходных металлов для объяснения свойств образуемых ими комплексов, следует помнить о приближенности водородоподобной модели строения атомов, которая в определенных ситуациях становится неадекватной. Что касается количественного согласия экспериментальных данных и квантово-химических рассчитанных свойств координационных соединений переходных металлов, то оно достигается лишь при достаточно точном учете энергии корреляции и релятивистских эффектов.
РЕЛЯТИВИСТСКИЕ ЭФФЕКТЫ
Скорости движения з возрастают с ростом порядкового номера атома пропорционально Z2. Например, скорость движения 1s – з (v) вокруг ядра с зарядом Z относительно скорости света (c) можно оценить по формуле
v/c = Z/137.
С ростом скорости движения з возрастает его масса (m), связанная с массой покоя (m0) известным уравнением:
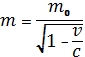
Например, масса электрона на s-орбитали ртути больше его массы покоя приблизительно на 20 %. Возрастание массы, как видно из формулы для радиуса первой боровской орбиты Q0 = 4рh2/mZ2e2 приводит к уменьшению радиуса орбиты. Наибольшее (и близкое по величине) уменьшение радиуса имеют s - и p1/2 - орбитали. Сжатие p2/3 - орбитали значительно меньше.
Более компактное расположение s-, p - з вокруг ядра вызывает возрастание их экранирующего действия. Это в свою очередь приводит к дестабилизации d-, f-орбиталей, они становятся более диффузными. Таким образом действие релятивистского эффекта на d- и f-орбитали непрямое и вызывается усилением экранирующего действия s-, p – з. Рассчитанные величины релятивистской стабилизации 6s-, 6p-орбиталей и дестабилизации 5d-орбитали атома золота показаны на рис. …
Рассчитанные энергии АО атома золота с учетом (Р) и без учета (НР) релятивистского эффекта. |
Квантово-химические расчеты потенциалов ионизации и электросродства тяжелых атомов дают существенно лучшее совпадение с экспериментом при учете релятивистских эффектов (р. э.)
Релятивистский эффект проявляется также в усилении магнитных взаимодействий, в частности наиболее сильного из них – спин-орбитального взаимодействия, взаимодействия собственного (спинового) магнитного момента электрона с его орбитальным моментом. Это взаимодействие не влияет на химические свойства легких атомов (первая треть элементов Периодической системы).
Спин-орбитальное взаимодействие проявляется в расщеплении водородо-подобных энергетических уровней атома Enl в соответствии со значениями квантового числа полного количества момента движения з – j. Рассел-саундовские термы ELS также расщепляются в соответствии со значениями j. Например, 5d-орбитали золота под действиемспин-орбитального взаимодействия расщепляются на 5d3/2 и 5d5/2.
Экспериментально установлено, что в рядах переходных металлов IV, V, VI периодов, лантаноидов и актиноидов радиусы атомов не только не растут, но даже уменьшаются. Это d-сжатие, лантаноидное сжатие.
Возникновение этого эффекта (сжатия) связывают с двумя факторами. Заполнение d- и f-орбиталей значительно меньше влияет на экранирование заряда ядра, чем заполнение s- и p-орбиталей. Более медленный рост эффективного заряда ядра в сравнении с ростом количества d-, f-электронов приводит к уменьшению радиусов атомов в этом ряду. Этот фактор называют орбитальным. Релятивистские эффекты стабилизируют внешние s- и p-орбитали, что также приводит к уменьшению радиусов атомов. Таким образом, оба фактора, орбитальный и релятивистский, действуют в одном направлении. Квантово-химические расчеты средних радиусов атомов показывают, что вклад орбитального сжатия оказался в несколько раз больше, чем релятивистского.
Ионные радиусы в ряду лантаноидов (от La до Lu) в комплексах с КЧ = 8 уменьшаются на 18,3 нм. 10 % этой величины обусловлено релятивистским эффектом.
Соединение атомов с образованием химического соединения целесообразно рассматривать как процесс, состоящий из нескольких этапов. Энергию образования комплекса MLn можно представить как сумму трех составляющих:
1) энергия возбуждения центрального атома, ДE1: M + ДE1 = M*;
2) энергия возбуждения лиганда, ДE2: L + ДE2=L*;
3) энергия взаимодействия M* с L,* ДE3: M* + L* = ML + ДE3.
Две первые составляющие определяют потери энергии на перевод свободных центрального атома и лаганда из основного состояния в валентное. Например, часто используют представления о валентных состояниях углерода sp3, sp2 и sp, характеризующих строение валентной оболочки углерода в алканах, алкенах и алкинах, соответственно. Образование этих состояний из основного состояния углерода (s2p2) требует затраты энергии.
Валентные состояния атомов переходных металлов характеризуются перераспределением электронов по ns-, np - и (n-1)d-орбиталям и образованием ионов.
В соответствии с этим, энергия образования (диссоциации) соединения зависит от величин этих составляющих – энергий образования валентных состояний центрального атома и лигандов.
Третья составляющая характеризует экзотермический процесс – взаимодействие центрального атома с лигандами, находящимися в валентном (возбужденном) состоянии.
Для образования соединения должно выполняться соотношение:
|ДE3| > |ДE1| + |ДE2|.
КВАНТОВО-ХИМИЧЕСКИЕ ПОЛУЭМПИРИЧЕСКИЕ ТЕОРИИ
В настоящее время для описания электронного строения и химической связи в координационных соединениях обычно используют три основные квантово-механические теории: теорию валентных связей (ТВС), теорию кристаллического поля (ТКП) и теорию поля лигандов (модифицированную теорию кристаллического поля МТКП, МОЛКАО).
Теория валентных связей была популярна в 1960-1970 гг. скорее из-за ее наглядности, а не из-за критериев точности и надежности получаемых результатов. Частично указанные достоинства обусловлены кажущейся близостью качественного аспекта теории и классическим представлением.
Теория валентных связей
В 1927 г. Гайтлер и Лондон на основе квантово-механического расчета молекулы водорода обосновали гипотезу Льюиса об образовании ковалентной химической связи посредством пары электронов с антипараллельными спинами. Полинг и Слэтер распространили эту концепцию на многоатомные молекулы с ковалентными связями. Развитый в их работах метод получил название метода валентных связей – ГЛСП (Гайтлер – Лондон – Слэтер – Полинг, 1927 г.) .
В ТВС октаэдрические комплексы описываются следующим образом:
1). Каждый лиганд считается донором, способным передавать пару з иону металла.
2). Ион металла должен располагать 6 свободными эквивалентными между собой у-орбиталями. Такой набор орбиталей можно получить из атомных spd орбиталей одним единственным способом – путем гибридизации s, px, py, pz, ![]() и
и ![]() .
.
Условием гибридизации является один и тот же класс симметрии и близость энергии орбиталей.
Число орбиталей центрального атома, участвующих в образовании координационных связей, равно числу лигандов d2sp3.
Гибридные орбитали имеют вполне определенное направление в пространстве.
Геометрическое строение и тип гибридизации комплексных ионов
КЧ | Тип гибридизации | Пространственное строение | Пример |
2 3 4 4 5 5 6 7 8 | sp sp2 sp3 dsp2 dsp3, d3sp d2sp 2, d 4s d2 sp3 d3sp3 d4sp3 | Линейное Тригональное Тетраэдр Квадрат Тригональная бипирамида Квадратная пирамида Октаэдр Пентагональная бипирамида Квадратная антипризма | [Ag(NH3)2] [HgI3] [FeBr4] [Cu Cl5] [Ni(CN)5] [Co(NH3)6] [V(CN)7] [TaF8] |
Рассмотрим с этих позиций строение некоторых комплексов:
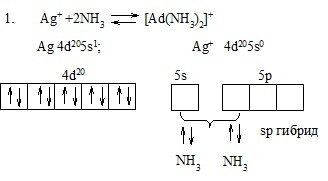
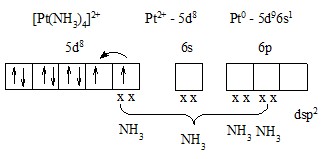
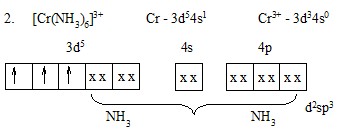
Эта диаграмма содержит следующую информацию:
1) чисто качественно указывает, что энергия валентных орбиталей убывает в последовательности
4p > 4s > 3d;
2) в комплексе существуют гибридные d2sp3-орбитали, занятые парами з от 6NH3;
3) на трех d- находятся три неспаренных з, спины которых параллельны. За счет 3 неспаренных з это соединение должно быть парамагнитным.
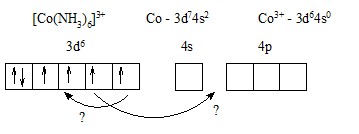
Два варианта: а) 2з промотировать на 4d или 5s;
б) поместить 2з на уже занятые 3d-орбитали. При этом нужно дважды затратить энергию на спаривание з.
Второй вариант энергетически более выгоден.
Тогда
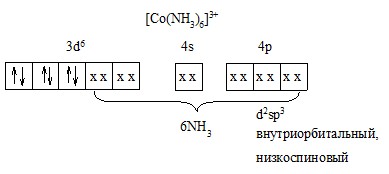
4) В [Co(NH3)6]![]() нет неспаренных з, комплекс диамагнитный. Все комплексы Co
нет неспаренных з, комплекс диамагнитный. Все комплексы Co![]() – диамагнетики.
– диамагнетики.
5). Исключение [CoF6]![]() – парамагнетик и его магнитный момент соответствует четырем неспаренным электронам.
– парамагнетик и его магнитный момент соответствует четырем неспаренным электронам.
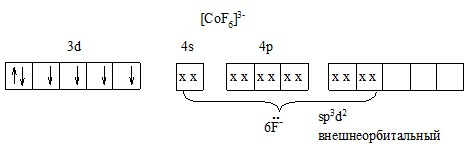
Еще одно предположение: гибридные d2sp3 образуются с участием так называемых внешних 4d-орбиталей, а 3d-орбитали при этом не затрачиваются.
Все октаэдрические комплексы Ni(II), имеющего восемь d з, за исклю- чением одного случая – парамагнитны.
Ni 3d24s2
Ni![]() 3d24s0
3d24s0
Высокоспиновые комплексы стали называть внешнеорбитальными, а низкоспиновые – внутриорбитальными.
МАГНИТНЫЕ СВОЙСТВА (Шрайвер)
Экспериментально низко - и высокоспиновые комплексы различаются по их магнитным свойствам.
Диамагнитные – выталкиваются.
Парамагнитные – втягиваются.
Степень парамагнетизма, как правило, выражают с помощью величины их магнитного дипольного момента: чем больше магнитный дипольный момент, тем больше парамагнетизм.
В современной технике – сверхпроводящее квантовое интерференционное устройство (SQUID, СКВИД).
В свободном ионе как орбитальный, так и спиновый угловые моменты приводят к возникновению магнитного момента и вносят вклад в парамагнетизм. Когда атом или ион является частью комплекса, то, благодаря взаимодействию электронов центрального иона с их несферическим окружением, орбитальный магнитный момент может быть исключен (говорят, что он погашен).
Однако спиновый угловой момент электрона существует и вызывает спиновый парамагнетизм, характерный для многих комплексов металлов.
Спиновый магнитный момент м комплекса с суммарным квантовым числом s равен
м = 2{s(s + 1)}1/2 мВ
L = l1+ l2, l1+l2-1, …, |l1 - l2|, S = s1+ s2, s1+s2-1, …, |s1-s2| ряд Клебша – Гордона, ![]()
мВ – магнетон Бора
мВ = ![]()
Его величина составляет 9,274·10-24 Дж/Тл.
![]()
где s – абсолютная величина спинового квантового числа,
g – гиромагнитное отношение (g – фактор).
g – для свободного з равна 2,00023 ![]()
![]() 2,00.
2,00.
![]()
![]() – спиновый угловой момент.
– спиновый угловой момент.
Спиновый магнитный момент одного з ![]()
![]()
Поскольку каждый неспаренный з имеет спиновое квантовое число Ѕ, mS = ЅN, где N – число неспаренных электронов, следовательно
м = {N(N + 2)}1/2 мВ =![]()
![]() .
.
Магнитные измерения можно использовать чтобы отличить низкоспиновые комплексы от высокоспиновых
![]() – (N = 4; м = 4,90 мB) – высокоспиновые,
– (N = 4; м = 4,90 мB) – высокоспиновые,
![]() – (S = 0; м = 0) – низкоспиновые.
– (S = 0; м = 0) – низкоспиновые.
Чисто спиновые значения магнитных моментов для разного числа
неспаренных электронов
Спиновые магнитные моменты
Ион | N | S | мs, мb | |
Расчет | Эксперимент | |||
Ti3+ V3+ Cr3+ Mn3+ Fe3+ | 1 2 3 4 5 6 | 1/2 1 3/2 2 5/2 3 | 1,73 2,83 3,87 4,90 5,92 | 1,7 – 1,8 2,7 – 2,9 3,8 4,8 – 4,9 6,3 |
Может быть еще и вклад орбитальной составляющей, например, d5 – низкоспиновые и 3d6 – высокоспиновые.
Пример. Определение электронной конфигурации из магнитных данных. Магнитный момент октаэдрического комплекса Co(II) составляет 4,0 мB.
Какова его электронная конфигурация?
Комплекс Co(II) представляет собой систему d7. Возможны две конфигурации: ![]() (высокоспиновая) – три неспаренных з;
(высокоспиновая) – три неспаренных з;
![]() (низкоспиновая) – один неспаренный з.
(низкоспиновая) – один неспаренный з.
Спиновые составляющие магнитного момента, рассчитанные по формуле, соответственно равны 3,87 мB и 1,73 мB.
Данные эксперимента совпадают с расчетными данными. Значит электронная конфигурация будет ![]() .
.
МВС хорошо описывает геометрическое строение комплексов и объясняет их магнитные свойства. Однако этот метод не позволяет оценить, какой из двух возможных вариантов заполнения орбиталей электронами предпочтительней, не дает возможности объяснить различную окраску комплексных соединений.
