
Партнерка на США и Канаду по недвижимости, выплаты в крипто
- 30% recurring commission
- Выплаты в USDT
- Вывод каждую неделю
- Комиссия до 5 лет за каждого referral

Пропуск экзона в гене миостатина при помощи антисмысловых олигонуклеатидов приводит к гипертрофии мышц у мышей после лечения октагуанидином морфолино олигомерами.
Ряд стратегий были предложены для повышения мышечной массы и силы при лечении ряда возрастных мышечных расстройств и различных нервно-мышечных расстройств, включая мышечные дистрофии. Миостатин, трансформирующий фактор роста-в, который также называется рост и дифференцировку, является негативным регулятором роста мышц, и ось регуляции миостатина была одним из основных направлений в такой стратегии. Отсутствие миостатина обуславливает значительное увеличение массы скелетных мышц, чем у животных дикого типа. Первая естественная мутация миостатина в организме человека также была определена у молодых мальчиков. Блокада миостатина предлагает стратегию противодействия повреждению мышц, включая дистрофию Дюшенна. Доставка ингибирующих миостатин генов, в том числе факторов роста и дифференцировки: сывороточного белка-1 (GASP-1), фоллистатин связанных генов (FLRG), фоллистатин-344 (FS) и пропептида миостатина через адено-связанный вирус приводит к увеличению мышечной массы у подопытных животных. Использование потенциально терапевтических антимиостатин-блокирующих антител высокой аффинности оказалось перспективной стратегией. Тем не менее, есть некоторые ограничения, связанные с использованием антител к миостатину, которые включают трудности с долгосрочной устойчивостью, нежелательными иммунными реакциями, а также ингибирующими эффектами точно не относящиеся к миостатину в отношении роста мышц. Значительное увеличение мышечной массы также наблюдались с помощью доставки адено-связанным вирусным вектором фрагмент гена рекомбинантного пропептида миостатина, или ретровирусом системы РНК-интерференции (RNAi) . Оба подхода имеют проблемы безопасности из-за возможныой генотоксичности при неконтролируемой вставке генома вектора в хромосомы, системы РНК-интерференции сталкиваются с дополнительными препятствиями в плане эффективной доставки молекул РНК-интерференции у моделей заболевания для клинических исследований. Терапия на основе РНК модуляции имеет потенциал, чтобы преодолеть трудности, с которыми сталкиваются обычные методы генной терапии. Антисмысловые олигонуклеотиды (АО) способны к гибридизации с последовательностью ДНК, ДНК гибрид не реагирует с РНКазой Н, что приводит к подавлению транскрипции гена. В альтернативном подходе, антисмысловая опосредованная модуляция пре-мРНК сплайсинга была использована впервые Dominski и Kole. В первых экспериментах АО были направлены на активацию сайтов сплайсинга в в-глобине (НВВ) и трансмембранном регуляторе проводимости при муковисцидозе (CFTR) для того, чтобы восстановить нормальный сплайсинг при в - талассемии и кистозном фиброзе. Идентификация экзон / интрон границ сплайсинга и, следовательно, включение экзонов в мРНК, как широко полагают, зависит от усилителя экзонного сращения (ESE). Маскируя эти ESE сайты с последовательностью оптимизированного АО, целевые экзоны больше не признаются экзонами, и сращены с соседними интронами. Это так называемый антисмысловой пропуск экзона, который уже используется в клинике, чтобы частично исправить мутировавший дистрофин, и конвертировать тяжелый фенотип миодистрофии Дюшенна в более мягкую мышечную дистрофию Беккера. Клинические испытания, чтобы определить профиль безопасности и эффективность однократного внутримышечного введения двух различных химических АО,, 2'-O-метил фосфоротиоат (2'OMePS) АО и фосфоротиамидат морфолино олигомеров (ПМ), проведены у пациентов с мышечной дистрофией в последнее время. Процедура хорошо перенесена всеми пациентами и введение АО индуцировало производство дистрофина. 2'OMePS АО, будучи заряжены отрицательно, легко доставлены в пробирке, в то время как в естественных условиях может наблюдаться более устойчивый эффект в связи с их устойчивостью к ферментативной дегенерации, и из-за их длинной последовательности увеличились близость к цели. Когда сопряженные с различными пептидными производными, или с дендримерным окта-гуанидином (так называемый Vivo-морфолино), они демонстрируют значительное увеличение поставки. Мы приняли этот подход с использованием АО с различными химическими модификациями, чтобы исследовать результаты блокады миостатина от пропуска экзона. Пропуск экзона 2 (374 нуклеотидов) миостатина, по прогнозам, приведет к сплайсингу вне рамки считывания экзонов 1 и 3, и блокада миостатина за счет усечения открытой рамки считывания и нонсенс-опосредованного распада мРНК. Данные, которые мы представляем здесь являются доказательством правильности принципа, что опосредованный антисмысловыми олигонуклеотидами пропуск экзона приводит к значительной физиологической блокаде миостатина в пробирке и в естественных условиях. Этот вид лечения может таким образом, потенциально являться частью эффективной стратегии по улучшению различных групп мышц, а вместе с восстановлением дистрофина или увеличением его уровня служить лечением мышечной дистрофии Дюшенна.
Резултаты
Биоинформационный анализ и проектирование конкретной АО для пропуска экзона 2 гена миостатина
Анализ экзона 2 миостатина был выполнен с помощью трех инструментов биоинформатики: ESE поиска, PESX, и ESE, для идентификации и обнаружения ESEs и супрессоров сплайсинга. Результаты этих алгоритмов показаны на рис 1а. Серии перекрывающих АО были разработаны и синтезированы 2'OMePSs и PMO, чтобы охватить последовательности, где кластеры ESE, которые были предсказаны одной или несколькими программами, совпадают.
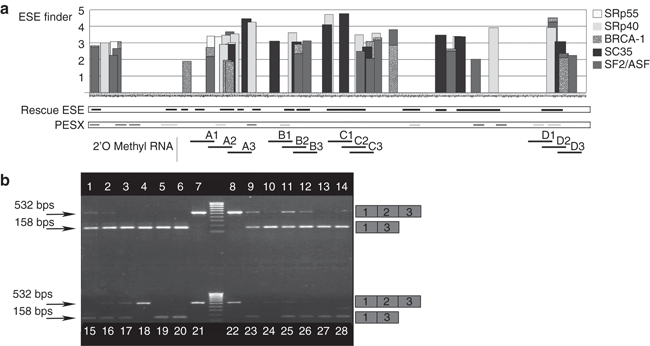
Высокий уровень пропуска экзона 2 миостатина в C2C12 культуре клеток после лечения целого ряда АО.
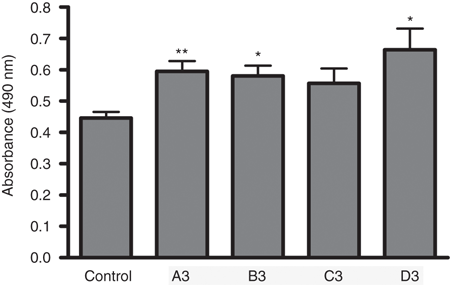
Для того, чтобы проверить эффективность этих последовательностей АО в C2C12 культуры мышечных клеток трансфицировали олигомеры 2'OMePS для пропуска экзонов в гене миостатина, контроль осуществляется ОТ-ПЦР по РНК, выделенной из трансфицированных и контрольных клеток. Результат горизонтального электрофореза в геле агарозы для разделения продуктов показан на рисунке 1b. Уровень пропуска, произведенный каждым АО на 250 нмоль / л был определен полуколичественным денситометрическим анализом. Все разработанные 2'OMePSs показали пропуск экзона 2 миостатина в C2C12 культурах, но на различных уровнях относительной эффективности. A2 и A3 индуцировали почти 100% пропуска; B3 (74%), C3 (41%) и D3 (48%) также вызывали значительный уровень пропуска. Характер предполагаемых антисмысловых продуктов exon1-exon3 сращивания был подтвержден с помощью секвенирования продуктов (данные не представлены).Антисмысловой пропуск экзона 2 и блокада миостатина приводит к увеличению C2C12 клеточной пролиферации. Для того, чтобы убедиться, что АО-опосредованной пропуск экзона 2 и блокада миостатина приведет к значительному биологическому ответу. Аутокринная активность миостатина на C2C12 пролиферацию клеток оценивали после лечения культур 2'OMePSs нацеленным на экзон 2. Анализ пролиферации клеток был основан на определении активности лактатдегидрогеназы метаболически активных клеток. Результаты анализа пролиферации ясно показали заметную разницу в клеточной пролиферации C2C12 клеток, обработанных АО к экзону 2, по сравнению с макетом трансфецированных контрольных клеток (рис. 2). Статистический анализ данных с использованием отдельных парных т-тестов показал, что олигомеры A3 (P = 0,0031), В3 (P = 0,0055) и D3 (P = 0,0115) вызвали значительное увеличение пролиферации клеток, по сравнению с группой контроля. Олигомеры C3 (P = 0,0534) не производят статистически значимых изменений.
Демонстрация пропуска экзонов у мышей после внутримышечной инъекции олигомеров 2'OMePS.
![]()
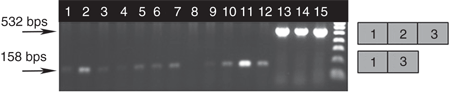
На основе результатов ОТ-ПЦР, полученные в лабораторных исследованиях, двух 2'OMePS олигомеров (A2 и B3) были выбраны для оценки их способности индуцировать эффективный пропуск экзонов в естественных условиях.2'OMePS олигомер (3 нмоль) вводят внутримышечно в переднюю большеберцовую (TA) мышцу мышей. Через две и четыре недели после инъекции мышцы были извлечены, их взвешивают, РНК экстрагировали и
![]()
анализировали на присутствие пропуска экзона 2 миостатина. Оба реагента (A2 и B3), индуцировали значительный уровень пропуска экзона 2 миостатина через 2 и 4 недели после одной инъекции 2'OMePS олигомера (рис. 3). ОТ-ПЦР анализ РНК был проведен с целью определить, какой из двух 2'OMePSs был более эффективным в естественных условиях. Олигомеры A2 дали 25,6% пропуска, и B3 дали 54,6% пропуска в точке 2 недели. Тем не менее, после 4 недель A2 дали 48,6%, тогда как пропуск B3 составил 24,5% . Несмотря на пропуск экзона 2 миостатина было очевидно, что эффект был не достаточным, чтобы увидеть значительные изменения в мышечной массе(данные не показаны). Из предыдущих работ пропуск экзонов для дистрофина, хорошо известно, что внутримышечные инъекции АО в неповрежденные мышцы не очень эффективны.
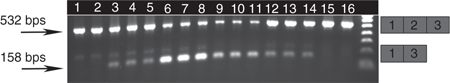
Высокий уровень пропуска экзона 2 миостатина в C2C12 культуре клеток после лечения РМО, разработанными на основе данных 2'OMePS.
Исследования на животных, проведенные выше, установили, что пропуск экзона гена миостатина наблюдается после внутримышечного введения 2'OMePS АО был недостаточным, чтобы вызвать изменение массы TA. Соединение PMO продемонстрировал очень высокую эффективность in vivo. Таким образом, РМО были разработаны на основе наиболее эффективного 2'OMePS АО (A3, B3, C3, D3) и первоначально протестированы в лабораторных условиях. РМО являются незаряженными химическими веществами и напрямую не взаимодействуют с поликатионными липофектаминами – реагентоми трансфекции. В целях обеспечения разумной эффективности трансфекции в C2C12 клетки РМО гибридизировали к дополнительным, так называемым, природным отрицательно заряженным олигонуклеотидам ДНК, как описано ранее. ОТ-ПЦР анализ мРНК из C2C12 клеток, обработанные PMO, показали, что пропуск экзонов был вызван во всех испытаниях (рис. 4).
Системное введение РМО конъюгированных с октагуанидином дендримером в результате дает пропуск экзона миостатина связанный со значительным увеличением мышечной массы и размера миофибрилл.
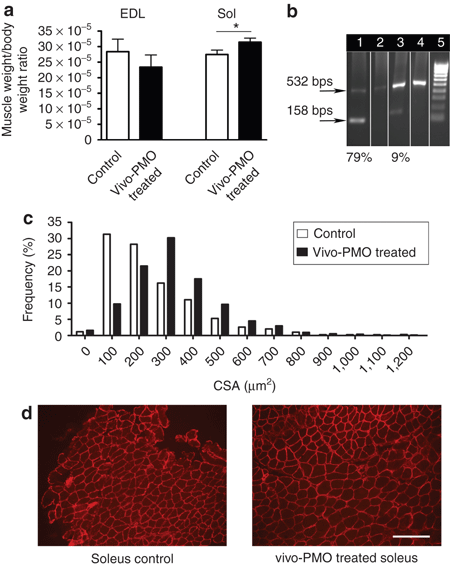
Сопряжение PMO с октагуанидином дендримером (так называемый Vivo-РМО) значительно увеличивает доставку и эффективность PMO направленых против экзона 23 гена дистрофина по сравнению с немодифицированной формой PMO. Таким образом, Vivo-PMO на основе последовательностей ранее проверенных 2'OMePS олигомеров был произведен для оценки системного внутрисосудистого режима лечения. Мышам вводили 6 мг / кг Vivo-PMO-D3 на пять еженедельных внутривенных инъекций, измерялись вес тела и масса ТА., камбаловидной и длинного разгибателя пальцев (EDL) через 10 дней после последней инъекции. Среди мышц, проанализированных у подопытных животных, камбаловидная мышца показала статистически значимое изменение массы (P = 0,034) (рис. 5а). В соответствии с этим, высокий уровень транскриптов с пропуском экзона 2 миостатина был продемонстрирован в камбаловидной мышце(79%), в то время как очень низкий уровень пропуска наблюдалася в EDL (9%) (рис. 5б). Важно отметить, что площадь поперечного сечения (CSA) мышечных волокон в камбаловидной мышце обработанных животных значительно увеличился (P <0,0001; средний уровень составил 254 ± 5 мкм2 для контроля и 333 ± 3 мкм2 для РМО-обработанных животных (n = 6) с значительным сдвигом в распределении (ч2 = 38,34; DF = 12). (рис. 5в, г), не наблюдалось изменение CSA в EDL мышце (данные не представлены).
Дискуссия
Было установлено, что миостатин является негативным регулятором скелетных мышц. Несколько подходов были использованы для блокады этого фактора, чтобы вызвать увеличение роста скелетных мышц. Использование АО вызвает пропуск экзона, и тем самым блокаду экспресиии миостатина, что представляет несколько преимуществ по сравнению с другими используемыми в настоящее время подходами генной терапии. Во-первых, не существует риска неконтролируемого введения в геном АО, как и в случае вирус опосредованных методов. Кроме того, в соответствующем режиме дозирования уровень пропуска экзона можно регулировать, и при необходимости лечение может быть прервано. Важно, что АО не производит каких-либо токсических эффектов или иммунного ответа у моделей животных, а также при использовании в клинических испытаниях. Здесь мы показываем, что 2'OMePS, разработанные с использованием алгоритмов, привели к существенному уровеню пропуска экзона 2 миостатина в пробирке. Миостатин, являющийся ингибитором миогенной дифференциации, контролирует распространение миобластов. Таким образом, блокада миостатина, как ожидается, вызовет увеличение пролиферации клеток. АО были биологически активными и вызывали повышение C2C12 клеточной пролиферации. Эффективность блокады при пропуске экзонов в естественных условиях оказалась более сложной, чем созданная в пробирке. Эффективность пропуска экзонов в гене миостатина была проверена путем введения 2'OMePS внутримышечно. Внутримышечное лечение одной мышцы индуцировало пропуск экзонов, но, как казалось, не влияет на деятельность миостатина. Это может быть связано с поставкой биологически активного миостатина из мышц в кровоток. Кроме того, в теории все тело должно получать лечение. По этим причинам, мы решили вводить АО с помощью системной инъекции в вену хвоста в наших дальнейших экспериментах. Состав PMO был выбран для эксперимента из-за его большей стабильности по сравнению с 2'OMePS, а также потому, что РМО, как сообщается, имеют более продолжительный эффект in vivo. Это особенно важно для блокады белков таких, как миостатин, которые не имеют длительного периода полувыведения. Для того, чтобы достичь разумного эффекта в неповрежденной мышце должны использоваться PMO конъюгированные с системой доставки. Vivo-PMO является коммерчески доступным и, как сообщается, может быть эффективным у нормальных здоровых мышей. После введения Vivo-PMO было получено существенное увеличение размера мышц и изменение размера волокна CSA, но только в камбаловидной мышце. Дифференциальный отклик в EDL и камбаловидной мышцах может быть отчасти из-за большего количества ActRIIB экспрессирующихся на поверхности EDL мышцы, или потому, что внутренний уровень миостатина больше в быстрых (миозина типа IIb положительный) волокнах. Альтернативно, мы можем предположить, что использованный режим дозирования, как сообщается, будет оптимальным для Vivo-PMO для пропуска экзонов в гене дистрофина, не даст достаточной пропуск в гене миостатина в EDL. В случае пропуска экзонов в гене дистрофина период полураспада белка дистрофина и мРНК очень велик и, следовательно, относительно меньшие
![]()
дозы АО дают более устойчивый результат. Однако, при пропуске в гене миостатина это, пожалуй, потребует более частого введения, чтобы иметь более устойчивое присутствие АО. Это может объяснить слабый эффект с точки зрения целого тела, что наблюдается в естественных условиях. Интересно, что только камбаловидная мышца показала значительное увеличение веса и размера волокна. Это согласуется с некоторыми ранее опубликованными данными, показывающими, что камбаловидная мышца является наиболее пораженной мышцей при системном подходе к блокаде миостатина. Наши результаты представляют собой доказательства правильности принципа, что блокада миостатина может быть получена пропуском экзона из транскриптов с помощью АО. Различные маршруты доставки, режимы дозирования и / или АО последовательности должны быть исследованы в будущих исследованиях для обеспечения эффективной в естественных условиях блокады экспрессии миостатина для максимального терапевтического эффекта. Эта работа имеет потенциал для разработки эффективного лечения для ряда возрастных мышечных расстройств и различных нервно-мышечных расстройств, включая мышечные дистрофии.
Ссылки
McPherron, AC, Lawler, AM and Lee, SJ (1997). Regulation of skeletal muscle mass in mice by a new TGF-в superfamily member. Nature 387: 83–90. | Article | PubMed | ISI | ChemPort | Schuelke, M, Wagner, KR, Stolz, LE, Hьbner, C, Riebel, T, Kцmen, W et al. (2004). Myostatin mutation associated with gross muscle hypertrophy in a child. N Engl J Med 350: 2682–2688. | Article | PubMed | ISI | ChemPort | Bogdanovich, S, Perkins, KJ, Krag, TO, Whittemore, LA and Khurana, TS (2005). Myostatin propeptide-mediated amelioration of dystrophic pathophysiology. FASEB J 19: 543–549. | Article | PubMed | ChemPort | Foster, K, Graham, IR, Otto, A, Foster, H, Trollet, C, Yaworsky, PJ et al. (2009). Adeno-associated virus-8-mediated intravenous transfer of myostatin propeptide leads to systemic functional improvements of slow but not fast muscle. Rejuvenation Res 12: 85–94. | Article | PubMed | ChemPort | Qiao, C, Li, J, Jiang, J, Zhu, X, Wang, B, Li, J et al. (2008). Myostatin propeptide gene delivery by adeno-associated virus serotype 8 vectors enhances muscle growth and ameliorates dystrophic phenotypes in mdx mice. Hum Gene Ther 19: 241–254. | Article | PubMed | ChemPort | Foster, KW (2009). Eye evolution: two eyes can be better than one. Curr Biol 19: R208–R210. | Article | PubMed Haidet, AM, Rizo, L, Handy, C, Umapathi, P, Eagle, A, Shilling, C et al. (2008). Long-term enhancement of skeletal muscle mass and strength by single gene administration of myostatin inhibitors. Proc Natl Acad Sci USA 105: 4318–4322. | Article | PubMed | ChemPort | Wagner, KR, Fleckenstein, JL, Amato, AA, Barohn, RJ, Bushby, K, Escolar, DM et al. (2008). A phase I/IItrial of MYO-029 in adult subjects with muscular dystrophy. Ann Neurol 63: 561–571. | Article | PubMed | ChemPort | Whittemore, LA, Song, K, Li, X, Aghajanian, J, Davies, M, Girgenrath, S et al. (2003). Inhibition of myostatin in adult mice increases skeletal muscle mass and strength. Biochem Biophys Res Commun 300: 965–971. | Article | PubMed | ISI | ChemPort | Yang, Z, Zhang, J, Cong, H, Huang, Z, Sun, L, Liu, C et al. (2008). A retrovirus-based system to stably silence GDF-8 expression and enhance myogenic differentiation in human rhabdomyosarcoma cells. J Gene Med 10: 825–833. | Article | PubMed Dumonceaux, J, Marie, S, Beley, C, Trollet, C, Vignaud, A, Ferry, A et al. (2010). Combination of myostatin pathway interference and dystrophin rescue enhances tetanic and specific force in dystrophic mdx mice. Mol Ther 18: 881–887. | Article | PubMed Weinstein, S and Peer, D (2010). RNAi nanomedicines: challenges and opportunities within the immune system. Nanotechnology 21: 232001. | Article | PubMed Hausen, P and Stein, H (1970). Ribonuclease H. An enzyme degrading the RNA moiety of DNA-RNA hybrids. Eur J Biochem 14: 278–283. | Article | PubMed | ISI | ChemPort | Zamecnik, PC and Stephenson, ML (1978). Inhibition of Rous sarcoma virus replication and cell transformation by a specific oligodeoxynucleotide. Proc Natl Acad Sci USA 75: 280–284. | Article | PubMed | ChemPort | Dominski, Z and Kole, R (1993). Restoration of correct splicing in thalassemic pre-mRNA by antisense oligonucleotides. Proc Natl Acad Sci USA 90: 8673–8677. | Article | PubMed | ChemPort | Friedman, KJ, Kole, J, Cohn, JA, Knowles, MR, Silverman, LM and Kole, R (1999). Correction of aberrant splicing of the cystic fibrosis transmembrane conductance regulator (CFTR) gene by antisense oligonucleotides. J Biol Chem 274: 36193–36199. | Article | PubMed | ISI | ChemPort | Sierakowska, H, Sambade, MJ, Agrawal, S and Kole, R (1996). Repair of thalassemic human в-globin mRNA in mammalian cells by antisense oligonucleotides. Proc Natl Acad Sci USA 93: 12840–12844. | Article | PubMed | ChemPort | Dunckley, MG, Manoharan, M, Villiet, P, Eperon, IC and Dickson, G (1998). Modification of splicing in the![]()
![]()
Оригинал статьи: http://www. /mt/journal/vaop/ncurrent/full/mt2010212
Переведено проектом МОЙМИО:
![]()
http://www.mymio.org
