

Партнерка на США и Канаду по недвижимости, выплаты в крипто
- 30% recurring commission
- Выплаты в USDT
- Вывод каждую неделю
- Комиссия до 5 лет за каждого referral
УТВЕРЖДЕНЫ
Решением Совета
Евразийской экономической комиссии
от 20 г. №………………
Т Р Е Б О В А Н И Я
к внедрению, поддержанию и оценке системы менеджмента качества медицинских изделий в зависимости
от потенциального риска применения
I. Общие положения
1. Настоящие требования разработаны в соответствии с пунктом 2 статьи 31 Договора о Евразийском экономическом союзе от 29 мая
2014 года, пунктом 1 статьи 6 Соглашения о единых принципах и правилах обращения медицинских изделий (изделий медицинского назначения и медицинской техники) в рамках Евразийского экономического союза от 01.01.01 года и устанавливают в рамках Евразийского экономического союза (далее – Союз) требования к внедрению, поддержанию и оценке системы менеджмента качества медицинских изделий в зависимости от потенциального риска применения (далее – Требования).
Настоящие требования вступают в силу с момента принятия. Со дня вступления в силу настоящих Требований и до 1 января 2018 года действует переходный период, в течение которого оценка системы менеджмента качества производителей медицинских изделий в соответствии с настоящими Требованиями не проводится. В течение указанного переходного периода производитель при подаче заявления на регистрацию медицинского изделия классов потенциального риска применения 2а, 2б и 3 подает в составе регистрационного досье документы, подтверждающие наличие у него условий производства заявленного к регистрации медицинского изделия, соответствующих требованиям национального законодательства государства, резидентом которого он является (при наличии таких требований) и (или) копии сертификатов соответствия системы менеджмента качества требованиям ISO 13485, ГОСТ ISO 13485 или соответствующего национального стандарта.
Производители медицинских изделий, зарегистрированных до
1 января 2018 года, обязаны подтвердить внедрение системы менеджмента качества в соответствии с настоящими Требованиями в течение трех лет с даты выдачи регистрационного удостоверения.
2. Под системой менеджмента качества понимается организационная структура, функции, процессы, процедуры и ресурсы, необходимые для скоординированной деятельности по руководству и управлению организацией, являющейся производителем медицинских изделий, применительно к качеству.
3. Для целей настоящих Требований используются следующие понятия:
«аудит» – систематический, независимый и документированный процесс получения записей, фиксирования фактов или другой соответствующей информации, а также их объективного оценивания с целью установления степени выполнения заданных требований.
«оценка системы менеджмента качества» – аудит производителя медицинских изделий в целях подтверждения результативности функционирования системы менеджмента качества и обеспечения соответствия выпускаемых в обращение изделий применимым к ним общим требованиям безопасности и эффективности медицинских изделий;
«программа аудита» – совокупность мероприятий по проведению одного или нескольких аудитов, запланированных на конкретный период времени и направленных на достижение конкретной цели.
«план аудита» – описание деятельности и мероприятий по проведению аудита.
«проверяемая организация» – организация, подвергающаяся аудиту.
«соответствие» – выполнение требования.
«несоответствие» – невыполнение требования.
«корректирующее действие» – действие, предпринятое производителем медицинских изделий, с целью устранения причины обнаруженного несоответствия;
«наблюдатель» – лицо, сопровождающее группу по аудиту, но не проводящее аудит.
II. Требования к внедрению системы менеджмента качества медицинских изделий
4. Производители медицинских изделий, кроме нестерильных медицинских изделий класса 1 потенциального применения, до подачи документов на регистрацию обязаны внедрить систему менеджмента качества (далее – СМК) в зависимости от класса потенциального риска применения.
Производители нестерильных медицинских изделий класса 1 потенциального риска применения вправе внедрить и поддерживать СМК.
В случае если производители нестерильных медицинских изделий класса 1 потенциального применения прошли оценку СМК в соответствии с настоящими Требованиями, то внесение изменений в регистрационное досье и (или) регистрационное удостоверение для таких медицинских изделий осуществляется без проведения экспертизы безопасности, качества и эффективности в уведомительном порядке на период действия заключения по результатам оценки СМК. Производитель такого медицинского изделия в течение 1 месяца со дня внесения изменений в документы представленного в рамках регистрации медицинского изделия регистрационного досье уведомляет уполномоченный орган государства – члена, выдавший регистрационное удостоверение медицинского изделия, о внесении соответствующих изменений.
5. СМК производителя медицинских изделий должна соответствовать требованиям межгосударственного стандарта из перечня стандартов, в результате применения которых на добровольной основе полностью или частично обеспечивается соблюдение соответствия медицинского изделия Общим требованиям безопасности и эффективности медицинских изделий (далее – Общие требования). СМК должна обеспечивать соответствие выпускаемых в обращение медицинских изделий применимым к ним Общим требованиям.
6. Производители медицинских изделий класса 1, выпускаемых в обращение в стерильном виде, а также всех медицинских изделий класса 2а потенциального риска применения, могут исключить из области применения СМК процессы проектирования и разработки. Производители медицинских изделий классов 2б и 3 потенциального риска применения, должны иметь в области применения СМК процессы проектирования и разработки.
7. В случае передачи каких-либо процессов СМК третьим сторонам, производитель должен определить требования к этим процессам и обеспечить контроль за их выполнением. При этом производитель несет полную ответственность за передаваемые третьим сторонам процессы, а также качество производимого изделия. Производитель должен указать в структуре СМК такие третьи стороны и выполняемые ими процессы.
8. Внедрение СМК должно быть проведено производителем до завершения процедуры регистрации. В отношении образцов медицинских изделий, используемых для проведения испытаний в целях регистрации, производитель должен провести анализ и документировать их соответствие серийному выпуску изделий в рамках внедренной СМК.
9. Внедрение СМК должно быть подтверждено производителем медицинских изделий посредством включения в регистрационную документацию отчета по оценке СМК, выполненной любой уполномоченной организацией по оценке СМК государств – членов Союза. Для нестерильных изделий класса 1 внедрение СМК подтверждается включением в регистрационную документацию перечня внедренных процедур системы качества, а также последнего по времени отчета по внутреннему аудиту СМК.
III. Требования к поддержанию системы менеджмента качества медицинских изделий
10. Производители медицинских изделий, внедрившие СМК в соответствии с разделом II настоящих Требований, должны поддерживать ее в актуальном состоянии и обеспечивать ее результативность.
11. В рамках СМК производителя должна быть установлена документированная процедура мониторинга изменений, вносимых в регистрационную документацию на медицинское изделие, а также изменений в перечне стандартов, в результате применения которых на добровольной основе полностью или частично обеспечивается соблюдение соответствия медицинского изделия Общим требованиям.
В рамках СМК производитель должен хранить документы и записи, подтверждающие соответствие качества выпускаемых изделий, с учетом их назначенного срока службы или срока годности, но не менее 15 лет для имплантируемых изделий, и не менее 5 лет для любых других изделий.
12. Поддержание СМК должно быть подтверждено проведением ежегодной оценки, выполненной любой уполномоченной организацией по оценке СМК государств – членов Союза, с оформлением соответствующего отчета по оценке. Для нестерильных изделий класса 1 поддержание СМК подтверждается своевременной актуализацией внедренных процедур системы качества, ведением записей по менеджменту риска и ежегодным проведением внутреннего аудита СМК с оформлением соответствующего отчета.
IV. Требования к оценке системы менеджмента качества медицинских изделий
13. Оценку внедрения и поддержания СМК производителей медицинских изделий осуществляют Уполномоченные органы государств-членов Союза и (или) уполномоченные организации по оценке СМК в форме проведения аудита. Аудит СМК должен проводиться на соответствие требованиям стандарта на СМК, включенного в перечень стандартов, в результате применения которых на добровольной основе полностью или частично обеспечивается соблюдение соответствия медицинского изделия Общим требованиям.
Оценку внедрения и поддержания СМК производителей медицинских изделий класса 3 потенциального риска применения должны осуществлять только сами Уполномоченные органы государств-членов Союза или подведомственные им уполномоченные организации по оценке СМК.
14. Уполномоченные организации по оценке СМК должны быть аккредитованы в национальных системах аккредитации государств – членов Союза и иметь стандарт, указанный в п.13, в своей области аккредитации. Перечень уполномоченных организаций по оценке СМК с учетом конкретных групп и подгрупп медицинских изделий определяют Уполномоченные органы государств – членов Союза.
При наделении организаций по оценке СМК соответствующими полномочиями Уполномоченные органы государств-членов Союза должны руководствоваться документами IMDRF/MDSAP WG/N3 и WG/N4 или идентичными этим документам национальными стандартами.
15. Деятельность по оценке СМК должна планироваться. При составлении плана аудита СМК уполномоченными организациями должны учитываться все территориально обособленные производственные площадки производителя, а также перечень всех привлекаемых третьих сторон в соответствии с п.7 настоящих Требований. Производитель должен обеспечить возможность проверки уполномоченной организацией по оценке СМК всех значимых с точки зрения менеджмента риска процессов, переданных третьим сторонам. В случае если на одной производственной площадке производятся медицинские изделия, относящиеся к нескольким группам (подгруппам) медицинских изделий, согласно перечню групп, подгрупп медицинских изделий (Приложение ), то оценка СМК в рамках одного аудита может охватывать несколько групп (подгрупп) медицинских изделий в соответствии с заявкой производителя.
При планировании и проведении оценки СМК организации проводящие оценку должны руководствоваться соответствующими документами IMDRF/GHTF SG/3 и SG/4 или идентичными этим документам национальными стандартами.
16. Результаты оценки СМК должны распространяться на группу (подгруппу) медицинских изделий согласно Перечню групп, подгрупп медицинских изделий (Приложение ) в зависимости от класса риска выпускаемых медицинских изделий. Для медицинских изделий классов потенциального риска применения 1 и 2а результаты оценки СМК распространяются на группы медицинских изделий. Для медицинских изделий классов риска 2б и 3 результаты оценки СМК распространяются на подгруппы медицинских изделий.
17. Расходы по проведению оценки внедрения и поддержания СМК несет производитель по договору с уполномоченной организацией по оценке СМК. Тарифы на проведение оценки системы менеджмента качества могут устанавливаться Уполномоченными органами государств-членов Союза на основе нормативной продолжительности проверок согласно Порядку определения нормативной продолжительности оценки СМК производителей медицинских изделий (Приложение ).
18. Деятельность уполномоченных организаций по оценке СМК подлежит надзорным проверкам Уполномоченным органом не реже, чем один раз в два года на предмет соблюдения настоящих Требований. Уполномоченные органы должны размещать на своем информационном ресурсе в сети Интернет график проверок уполномоченных организаций по оценке СМК, в том числе с целью их уполномочивания, доступный для всех Уполномоченных органов государств-членов, не позднее чем за три месяца до начала ближайшей проверки.
Полномочия организаций по оценке СМК могут быть аннулированы Уполномоченным органом, если в результате надзорной проверки будет установлено, что организация по оценке СМК не соблюдает положений настоящих требований.
Уполномоченные органы государств-членов Союза вправе за свой счет направить своего представителя для участия в оценке СМК производителя медицинских изделий в качестве наблюдателя.
19. По результатам проведенной оценки уполномоченная организация по оценке оформляет отчет с заключением о соответствии или несоответствии СМК производителя медицинского изделия настоящим Требованиям. Уполномоченные организации по оценке СМК обязаны представлять в электронной форме в Уполномоченный орган, от которого они получили полномочия на проведение оценки, отчеты по результатам оценки в срок не позднее 15 рабочих дней после завершения аудита.
Уполномоченный орган предоставляет по запросам указанную информацию другим Уполномоченным органам государств-членов в срок не позднее 10 рабочих дней после поступления запроса.
20. Уполномоченные организации по оценке СМК не должны выносить положительного заключения о соответствии, если внедренная производителем СМК не соответствует настоящим Требованиям или не поддерживается в актуальном состоянии. Несоответствия, выявленные по результатам оценки, должны быть устранены производителем в ходе проверки или в срок, установленный организацией по оценке СМК.
21. При несогласии производителя с отрицательным заключением или по выявленным несоответствиям он имеет право направить претензию в организацию по оценке СМК и в Уполномоченный орган, от которого она получила полномочия на проведение оценки, в срок не позднее 30 рабочих дней со дня получения копии отчета по результатам оценки СМК.
22. Если производитель не устраняет несоответствия или нарушает сроки устранения несоответствий, уполномоченная организация по оценке СМК незамедлительно информирует об этом факте Уполномоченный орган государства-члена, уполномочивший организацию на проведение оценки. Уполномоченный орган государства-члена вправе приостановить выпуск медицинского изделия в обращение на территории государства-члена до устранения выявленных несоответствий. Уполномоченные органы государств – членов Союза информируются о приостановке (возобновлении) выпуска медицинского изделия в обращение на территории государства-члена посредством информационной системы Союза в сфере обращения медицинских изделий.
ПРИЛОЖЕНИЕ № 1
к Требованиям к внедрению, поддержанию и оценке системы менеджмента качества медицинских изделий в зависимости
от потенциального риска их применения
Порядок определения нормативной продолжительности проведения оценки СМК производителей медицинских изделий
1. Нормативная продолжительность оценки применяется для определения стоимости проведения оценки СМК независимо от ее фактической продолжительности.
2. Нормативная продолжительность оценки исчисляется в человеко-днях на основе 8-часового рабочего дня. Продолжительность оценки включает в себя время, проведенное непосредственно в проверяемой организации, и время, проведенное вне проверяемой организации и затраченное на планирование, анализ документации и составление отчетов.
Если объект(ы) оценки расположен(ы) в другом городе по отношению к организации проводящей оценку, то нормативная продолжительность увеличивается на два дня.
Если производитель имеет несколько территориально обособленных производственных площадок или передает все или часть процессов СМК третьим сторонам, то нормативная продолжительность увеличивается на два дня за каждую дополнительную производственную площадку, а также третью сторону, которые входят в структуру СМК производителя.
3. В качестве основы для расчета нормативной продолжительности оценки используется фактическая численность персонала проверяемого объекта, участвующего в процессах оцениваемой СМК. Численность персонала с частичной занятостью учитывается путем ее конвертации в эквивалентную численность персонала с полной занятостью.
4. Нормативная продолжительность оценки внедрения и поддержания СМК производителя медицинских изделий приведена в таблице в зависимости от фактической численности персонала проверяемого объекта(ов):
Фактическая численность, человек | Продолжительность проверки с целью оценки внедрения системы менеджмента качества, чел.-дней | Продолжительность проверки с целью оценки поддержания системы менеджмента качества, чел.-дней |
5-9 | 3 | 2 |
10-19 | 4 | 3 |
20-49 | 6 | 4 |
50-99 | 7 | 5 |
100-199 | 8 | 6 |
200-499 | 9 | 7 |
500-999 | 10 | 8 |
1000-1999 | 11 | 9 |
2000-4999 | 12 | 10 |
более 5000 | 13 | 11 |
Предлагается рассмотреть следующую таблицу из документа Международного Форума по Аккредитации, где Стадия 1 – время на проверку предоставленных документов, Стадия 2 - время на аудит в организации.
IAF MD9:2011 Issue 1, Version 2 Application of ISO/IEC 17021 in ISO 13485
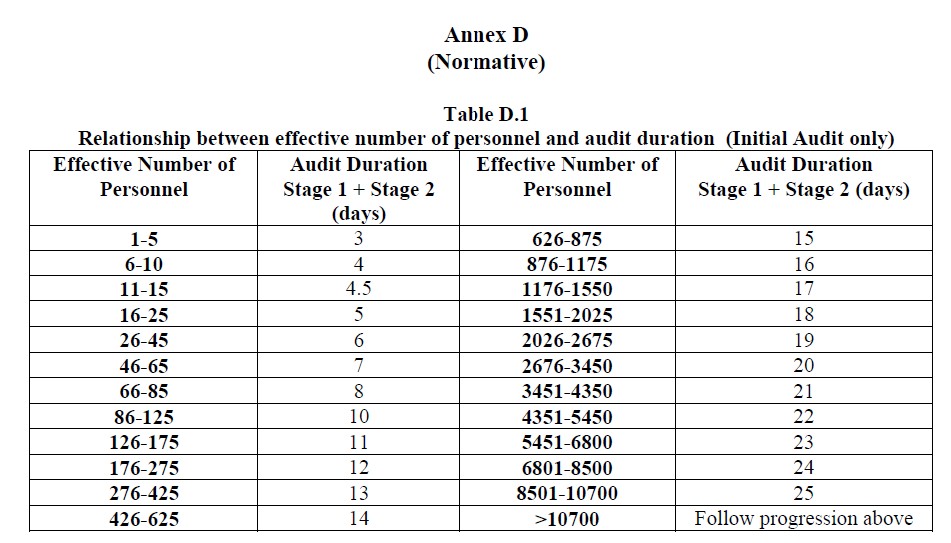
ПРИЛОЖЕНИЕ № 2
к Требованиям к внедрению, поддержанию и оценке системы менеджмента качества медицинских изделий в зависимости
от потенциального риска их применения
Перечень
групп, подгрупп медицинских изделий
Группы медицинских изделий (применяются для медицинских изделий классов риска 1, 2а) | Подгруппы медицинских изделий (применяются для медицинских изделий классов риска 2б, 3) |
1. Неактивные медицинские изделия (кроме изделий для in vitro диагностики) | 1.1. Неактивные сердечно-сосудистые имплантаты |
1.2. Неактивные ортопедические имплантаты | |
1.3. Неактивные имплантаты мягких тканей | |
1.4. Неактивные функциональные имплантаты | |
1.5. Неактивные зубные имплантаты и стоматологические материалы | |
1.6. Неактивные медицинские изделия для инъекций, вливаний, переливаний крови и диализа | |
1.7. Неактивные офтальмологические медицинские изделия | |
1.8. Неактивные ортопедические медицинские изделия и медицинские изделия для реабилитации | |
1.9. Медицинские изделия для контрацепции | |
1.10. Медицинские инструменты | |
1.11. Неактивные медицинские изделия для дезинфекции, гигиенической обработки и стерилизации медицинских изделий | |
1.12. Шовный материал, перевязочные средства и прочие неактивные медицинские изделия для лечения ран | |
1.13. Неактивные медицинские изделия, не вошедшие в подгруппы 1.1 -1.12 | |
2. Активные неимплантируемые медицинские изделия (кроме изделий для in vitro диагностики) | 2.1. Медицинские изделия для контроля физиологических показателей |
2.2. Медицинские изделия для визуализации, использующие ионизирующее излучение | |
2.3. Медицинские изделия для визуализации, не использующие ионизирующее излучение | |
2.4. Медицинские изделия для лучевой терапии, использующие ионизирующее излучение | |
2.5. Медицинские изделия для лучевой терапии, не использующие ионизирующее излучение | |
2.6. Медицинские изделия для литотрипсии | |
2.7. Активные медицинские изделия для экстракорпорального кровообращения, внутривенного вливания и афереза | |
2.8. Активные наркозно-дыхательные, гипербарические и медицинские изделия для респираторной терапии | |
2.9. Активные медицинские изделия для стимуляции и ингибирования | |
2.10. Активные хирургические медицинские изделия | |
2.11. Активные офтальмологические медицинские изделия | |
2.12. Активные стоматологические медицинские изделия | |
2.13. Активные медицинские изделия для дезинфекции и стерилизации медицинских изделий | |
2.14. Активные медицинские изделия для реабилитации и активные протезы | |
2.15. Активные медицинские изделия для позиционирования и перевозки пациентов | |
2.16. Самостоятельное медицинское программное обеспечение | |
2.17. Активные медицинские изделия для экстракорпорального оплодотворения и искусственного оплодотворения | |
2.18. Активные медицинские изделия, не вошедшие в подгруппы 2.1 -2.17 | |
3. Активные имплантируемые медицинские изделия | 3.1. Активные имплантируемые медицинские изделия для стимуляции и ингибирования |
3.2. Активные имплантируемые медицинские изделия для ввода лекарственных и иных веществ | |
3.3. Активные имплантируемые медицинские изделия, поддерживающие, замещающие или заменяющие функции организма | |
3.4. Радиоактивные имплантаты для внутритканевой лучевой терапии | |
3.5. Активные имплантируемые медицинские изделия, не вошедшие в подгруппы 3.1 - 3.4 | |
4. Медицинские изделия для диагностики in vitro | 4.1. Реагенты, наборы реагентов, калибровочные и контрольные материалы |
4.2. Приборы и оборудование для диагностики in vitro | |
4.3. Самостоятельное медицинское программное обеспечение для диагностики in vitro | |
4.4. Иные медицинские изделия для диагностики in vitro, не вошедшие в подгруппы 4.1 – 4.3 |
