

Партнерка на США и Канаду по недвижимости, выплаты в крипто
- 30% recurring commission
- Выплаты в USDT
- Вывод каждую неделю
- Комиссия до 5 лет за каждого referral

Системная генная терапия миодистрофии Дюшена у mdx мышей.
Michela Alessandra Denti,* Alessandro Rosa,* Giuseppe D’Antona,† Olga Sthandier,* Fernanda Gabriella De Angelis,* Carmine Nicoletti,‡ Mariacarmela Allocca,§ Orietta Pansarasa,† Valeria Parente,† Antonio Musarт,‡ Alberto Auricchio,§ Roberto Bottinelli,† and Irene Bozzoni*¶
*Institute Pasteur Cenci-Bolognetti, Department of Genetics and Molecular Biology and Institute of Molecular Biology and Pathology, University “La Sapienza,” P. le Aldo Moro 5, 00185 Rome, Italy;
†Department of Experimental Medicine, Human Physiology Unit, University of Pavia, Via Forlanini 6, 27100 Pavia, Italy;
§Telethon Institute of Genetics and Medicine (TIGEM), Via P. Castellino 111, 80131 Naples, Italy; and
‡Department of Histology and Medical Embryology, Centro di Eccellenza di Biologia e Medicina Molecolare and Interuniversity Institute of Myology, University of Rome “La Sapienza,” Via A. Scarpa 14, 00161 Rome, Italy
¶To whom correspondence should be addressed. E-mail: irene. [email protected]
Edited by Louis M. Kunkel, Harvard Medical School, Boston, MA, and approved January 6, 2006
Contributed by
Author contributions: M. A.D., A. R., G. D., F. G.D. A., A. M., A. A., R. B., and I. B. designed research; M. A.D., A. R., G. D., O. S., F. G.D. A., C. N., M. A., O. P., V. P., and A. A. performed research; M. A.D., A. R., G. D., O. S., F. G.D. A., A. M., A. A., R. B., and I. B. analyzed data; and M. A.D., A. R., G. D., F. G.D. A., A. M., A. A., R. B., and I. B. wrote the paper.
Received October 12, 2005
Резюме
Мышечная дистрофия Дюшена (МД) вызвана мутациями гена, ведущими к отсутствию белка дистрофина в поперечно - полосатых мышцах. Значительное количество этих мутаций связано с преждевременной остановкой кодонов. Многие такие мутации могут быть скорректированы на посттранскрипциональном уровне путем пропуска мутантного экзона. Мы получали такой эффект у mdx мышей при введении внутривенно аденоассоциированного вектора(ААВ) с антисмысловой последовательностью взятой из ядерной РНК U1 клеток. Системная доставка обеспечивала колонизацию по всему телу и существенное восстановление функциональных способностей in vivo и снижение креатинкиназы в сыворотке и уменьшение мышечного повреждения. Преобразование мышц из-за увеличения экспрессии дистрофина и восстановление функции стремились к нормальному значению. Этот подход обеспечивает прочное основание для системного применения AAV опосредованной передачи антисмыслового малого фрагмента ядерной РНК для лечения миодистрофии Дюшена.
Делеции и точечные мутации в человеческом гене дистрофина причина прогрессирующей миодистрофии Дюшена или более медленной миодистрофии Беккера. MDX мыши – это полезная модель для изучения эффективности лечения этих заболеваний. Существует две оспариваемые проблемы в генной терапии миодистрофии Дюшена: с одной стороны это тип лечебного гена и с другой - система его доставки для эффективного преобразования мышц. Из-за большого размера гена традиционное замещение гена провести достаточно сложно, только аденовирусные векторы могут доставить полную комплементарную ДНК. Однако опыты у мышей с использованием данных векторов показали наличие иммунного ответа на комплиментарную ДНК и трансформация мышц была недостаточно выраженной.
Были подходы в которых использовали доставку микрогена дистрофина адено-ассоциировенным вектором. Адено-ассоциированный вектор с капсидом ААV8 (или сходными, но не идентичными AAV1, или AAV6) в исследовании на WT мышах показал более эффективное преобразование мышц.
Недавно, был разработан метод коррекции фенотипа при миодистрофии Дюшена основанный на доставке антисмысловой последовательности, что приводит к пропуску экзона и лечение гентического повреждения на посттранскрипционном уровне.
Многие внутренние делеции, возникающие в регионе кодирования, а точнее в его обширной центральной области необязательно приводят к развитию прогрессирующей миопатии, при этом наблюдаются легкие симптомы миопатии. Поэтому для искусственного создания таких делеций было бы возможно включение специальных мутантных екзонов в матричную РНК, что позволит синтезировать короткий, но функциональный дистрофин.
Специфический пропуск экзона выполнен с использованием синтетического антисмыслового олигонуклеатида против специфической точки сплайсинга или экзонного энкансера in vitro, культивированного в культуре миобластлов человеческого и мышиного происхождения. В последствие, данный подход был использован в исследовании на mdx мышах при внутривенном или внутримышечном введении. Данный подход показал хорошие результаты и эффективное восстановление дистрофина, но ограниченный по времени эффект. Что вызвало рост интереса к данной методике. Стратегия заключалась в экспрессии этой последовательности как части стабильной клеточной РНК и, в частности, в основе РНК, обеспечивающей специфическую локализацию. В этом руководстве мы впервые описываем использование анти-нуклеотида U1 малой ядерной РНК (мяРНК) на миобластах человека с миодистрофией Дюшена с делецией 48-50 экзонов, для пропуска экзона 51. U7 мяРНК также была использована как основа антисмысловой экспрессии и возможность вызывать корректный пропуск экзона. Оба анти-нуклеотида способны вызывать преобразования в клетках млакопитающих и при введении в мышцы mdx мышей. Подобная эффективность при местном введении также обнаружена при использовании U1 мяРНК. В этой статье мы описали системное действие антисмысловых нуклеотидов, доставленных аден-ассоциированным вектором, при введении через вену хвоста mdx мышей.
Результаты
Мы заранее сообщаем, что пропуск экзона антисмысловой последовательностью против 5`и 3` участков сплайсинга экзона эффективен. В химическом синтезе создан U1≠23: конец 5` 8 нуклеотидов U1 мяРНК необходимые для распознования 5` сайтов сплайсинга пре-мРНК были заменены 54 участком комплементарной точки сплайсинга экзона 23 гена дистрофина. ААV2/1 эффективно доставляет антисмысловую конструкцию в мышцы. Для эффективной экспрессии антисмысловая последовательность конструируется в мышцах мышей in vivo, с использованием гибрида ААВ вектора, содержащего геном ААВ2 в капсиде ААВ1. Этот гибридный вектор полученный из ААВ серотипов изолирован для внутримышечного введения.
Копии производимые антисмысловой РНК могли распространяться в мышцах. 3-4Ч10^12 копий генома производных ААV (ААV-U1#23) введены в вену хвоста 6 недельных mdx мышей. Эта доза вектора считалась эффективной для преобразования мышечных волокон. В нашем исследовании мы не заметили разницы при введении одного вектора или совместном введении с VEGF. Инъекции были проведены 11мышам. Результаты оценивались через 6 и 12 недель после инъекции. У 6 различные ткани были протестированы на наличие EGFP. EGFP екрессия широко распространена у интактных животных.
![]()
Интенсивность флюоресценции была гетерогенной среди различных волокон. Интерсно, что в мышцах задних конечностей отмечено интенсивное преобразование, тогда как диафрагма и сердце показали менее однообразную экспрессию. EGFP-позитивные клетки были также найдены в печени, тогда как в других исследованных тканях не обнаружено трансгенной экспрессии. Наблюдаемые черты преобразования были схожи у различных животных, которым производили системное введение антисмысловой ААV конструкции.
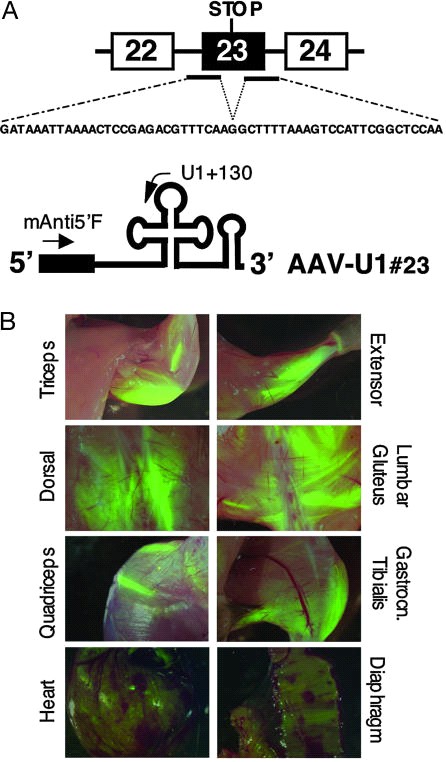
Для молекулярного анализа были резецированы отдельные мышечные волокна, для анализа РНК, белков и иммунофлюоресцентного анализа. Важно подчеркнуть, что образцы не были сопоставимы, поскольку имело место гетерогенное распределение ААV.
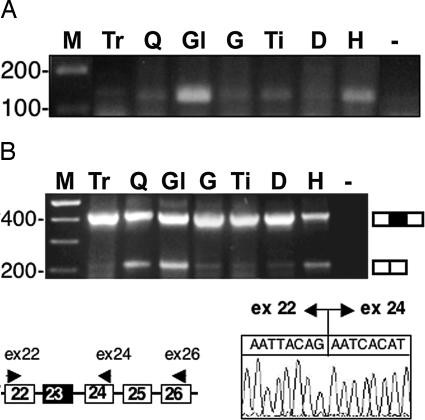
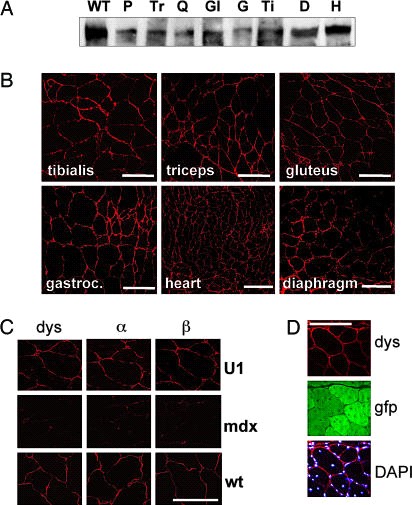
Экспрессия дистрофина устнавливалась с помощью Вестен блота общей экстракции белков. Рис. 3А - ААV-U1#23 конструкция освобожденная от синтеза дистрофина. Замечено, что пропуск одного экзона 23 может приводить к продукции белка короче на 73 аминокислоты, такое укорочение не может быть обнаружено у такого большого белка (427 кДа) в этом типе геля. Поскольку образцы РНК и белков получены из преобразованных и не преобразованных волокон, важно было опредилить уровень пропуска экзона и уровень дистрофина в отдельных волокнах.
Иммунофлеоресцентный анализ подтверждает, что AAV-U1#23 способен возобнавлять синтез дистрофина, который локализуется на периферии мышечных волокон. На рис. 3В показана иммунофлюоресценция дистрофина в образцах мышечных волокон полученных от mdx мышей, получавших AAV-U1#23. Синтез дистрофина был связан с нормальной экспрессией и локализацией б - и в-саркогликана, компонентов дистрофин-ассоциированного белкового комплекса. Иммуногистохимический анализ также показал соответствие между експрессией маркера EGFP и началом синтеза дистрофина.
На рис. 3D показана правельная локализация дистрофина в околомембранном пространстве. Микроскопический анализ показал гетерогенную колонизацию разных участков мышц, как уже показано на рисунке 1 при анализе на EGFP. Нет значительной разницы в колонизации и эффективности данного лечения у 6 недельных мышей.
Диафрагма представляет одну из самых поставленных под угрозу мышц как у mdx мышей, так и у человека, из-за большой функциональной нагрузки. Несмотря на низкие уровни экспрессии EGFP в диафрагме леченных мышей, мы наблюдали начало синтеза дистрофина. Подобные результаты были получены при исследовании сердца. Несмотря на то, что по ранее полученным данным сердце-орган, в котором трудно получить распространенную генную трансформацию; начало синтеза дистрофина, установленное в наших исследованиях, является обнадеживающим результатом.
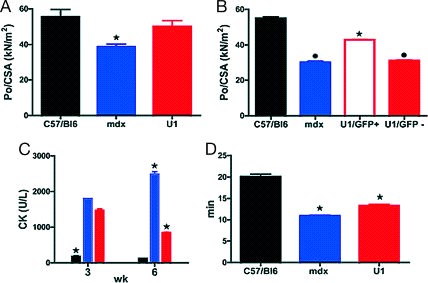
Для определения суммы экспрессии дистрофина мы анализировали силу сокращения
![]()
одиночного волокна, взятого из икроножной мышцы контрольной группы и группы мышей, получаюших лечение. Мы объединили 2 типа волокон 2Х и 2В, так как в таких мышцах это основные типы волокон, и не было разницы в силе и площади поперечного сечения между этими типами волокон. Сначала мы анализировали крупную популяцию (n=120, n=3 в каждой группе) мышечных волокон каждой группы. Самая низкая сила была в контрольной группе mdx мышей и приближалась к нормальной у мышей получавших лечение. В
![]()
подтверждение восстановления мышечной силы из-за начала синтеза дистрофина мы анализировали эти показатели у второй популяции мышц (n=150, n=3 для каждой группы) полученных из vastus от контрольной группы и mdx мышей, рассеченных под флюоресцентным микроскопом. Интересно, что EGFP-позитивные волокна были сильней, чем EGFP-негативные волокна. Результаты подтверждают, что экспрессия дистрофина у мышей получавших лечение вызывает восстановление мышечной силы.
Для подтверждения защиты мембраны волокон дистрофином, определялись уровни креатинкиназы в сыворотке через 3 и 6 недель после инъекции. Рис. 4С показывает, что системное введение AAV-U1#23 снижало уровень креатинкиназы.
Мы предполагали, ч то системное лечение способно улучшать функции мыщц у мышей in vivo. Через 6 недель после инъекции был проведен тест истощения на беговой дорожке.
Группы мышей, которые не получали лечения, показывали снижение толерантности к нагрузке, а мыши получавшие лечение, напротив показали улучшение работоспособности.
Обсуждение
При рассмотрении эффективности данного метода, который может быть использован для лечения большинства мутаций гена дистрофина, параллельные подходы сфокусированы на поиске возможностей преобразования мышц при помощи синтеза антисмысловой последовательности как части стабильной клеточной РНК. Данная стратегия была успешно использована при введении ААV c U7 мя РНК в мышцу или артерии нижней конечности. Однако, учитывая эффекты вектор-опосредованной генной терапии при миопатиях, возникает необходимость в доставке препарата ко всем мышцам, то есть в улучшении системных средств доставки. В этом исследовании мы рассмотрели системное использование химеры ААV вируса, восстановление дистрофина и дистрофин - ассоциированного комплекса во всем теле, и функциональное улучшение параметров мышечной силы после начала экспрессии вирусной РНК.
Значительные результаты были продемонстрированы при использовании двух схожих подходов: Исследование силы одиночного мышечного волокна in vitro и подвижности in vivo. Анализ удельной силы одиночного мышечного волокна очень чувствительный метод для выявления повреждений мышц у мышей, моделей миодистрофии Дюшена и функционального восстановления после лечения. Восстановление удельной силы отдельного мышечного волокна ясно показывает эффективность системного лечения. Важно отметить, что восстановление функции было непосредственно связано с восстановлением дистрофин-ассоциированного белкового комплекса. Причина по которой восстановление дистрофин-ассоциированного белково го комплекса могло сводиться к минимуму в покрытых кожей мышцах остается не ясна. Причин может быть несколько: с одной стороны это может быть гиперкатаболизм миофибрилл при активации кальций зависимых протеаз, с другой стороны распределение силы между мышцей и кожей. И действительно мы нашли нармальную локализацию дистрофина в препаратах покрытых кожей.
Конечно, восстановление силы мышц представляется целью любой терапии миодистрофии. Степень восстановления силы после 12 недельного лечения ободряет исследователей на продолжение исследовании данного метода. Полученные результаты клинически уместны и дают надежду на обнаружение способа лечения миодистрофии у людей с использованием данного метода..Наши данные подтверждают эффективность лечения поскольку введение вируса вызывало улучшение функциональных параметров, и поддержание продукции дистрофина в течение как минимум следующих 12 недель. Предположение о наличии иммунного ответа на введение вируса не подтвердились. Хотя у больших животных и людей из-за использования больших доз вектора скорее всего будет развиваться иммунный ответ. Разумная оценка риска и продуманные подходы к применению позволят привести к использованию данного метода у людей.
![]()
Список литературы.
1. Koenig M., Beggs A. H., Moyer M., Scherpf S., Heindrich K., Bettecken T., Meng G., Muller C. R., Lindlof M., Kaariainen H., et al. Am. J. Hum. Genet. 1989;45:498–506. [PMC free article] [PubMed]
2. Sicinski P., Geng Y., Ryder-Cook A. S., Barnard E. A., Darlison M. G., Barnard P. J. Science. 1989;244:1578–1580. [PubMed]
3. DelloRusso C., Scott J. M., Hartigan-O’Connor D., Salvatori G., Barjot C., Robinson A. S., Crawford R. W., Brooks S. V., Chamberlain J. S. Proc. Natl. Acad. Sci. USA. 2002;99:12979–12984. [PMC free article] [PubMed]
4. Jiang Z., Schiedner G., van Rooijen N., Liu C. C., Kochanek S., Clemens P. R. Mol. Ther. 2004;10:688–696. [PubMed]
5. Wang B., Li J., Xiao X. Proc. Natl. Acad. Sci. USA. 2000;97:13714–13719. [PMC free article] [PubMed]
6. Wang Z., Zhu T., Qiao C., Zhou L., Wang B., Zhang J., Chen C., Li J., Xiao X. Nat. Biotechnol. 2005;23:321–328. [PubMed]
7. Gregorevic P., Blankinship M. J., Allen J. M., Crawford R. W., Meuse L., Miller D. G., Russell D. W., Chamberlain J. S. Nat. Med. 2004;10:828–834. [PMC free article] [PubMed]
8. van Deutekom J. C., van Ommen G. J. Nat. Rev. Genet. 2003;4:774–783. [PubMed]
9. Aartsma-Rus A., Janson A. A., Kaman W. E., Bremmer-Bout M., van Ommen G. J., den Dunnen J. T., van Deutekom J. C. Am. J. Hum. Genet. 2004;74:83–92. [PMC free article] [PubMed]
10. Dunckley M. G., Manoharan M., Villiet P., Eperon I. C., Dickson G. Hum. Mol. Genet. 1998;7:1083–1090. [PubMed]
11. Mann C. J., Honeyman K., Cheng A. J., Ly T., Lloyd F., Fletcher S., Morgan J. E., Partridge T. A., Wilton S. D. Proc. Natl. Acad. Sci. USA. 2001;98:42–47. [PMC free article] [PubMed]
12. Lu Q. L., Mann C. J., Lou F., Bou-Gharios G., Morris G. E., Xue S. A., Fletcher S., Partridge T. A., Wilton S. D. Nat. Med. 2003;9:1009–1014. [PubMed]
13. Bremmer-Bout M., Aartsma-Rus A., de Meijer E. J., Kaman W. E., Janson A. A., Vossen R. H., van Ommen G. J., den Dunnen J. T., van Deutekom J. C. Mol. Ther. 2004;10:232–240. [PubMed]
14. Lu Q. L., Rabinowitz A., Chen Y. C., Yokota T., Yin H., Alter J., Jadoon A., Bou-Gharios G., Partridge T. Proc. Natl. Acad. Sci. USA. 2005;102:198–203. [PMC free article] [PubMed]
15. De Angelis F. G., Sthandier O., Berarducci B., Toso S., Galluzzi G., Ricci E., Cossu G., Bozzoni I. Proc. Natl. Acad. Sci. USA. 2002;99:9456–9461. [PMC free article] [PubMed]
16. Brun C., Suter D., Pauli C., Dunant P., Lochmuller H., Burgunder J. M., Schumperli D., Weis J. Cell. Mol. Life Sci. 2003;60:557–566. [PubMed]
17. Goyenvalle A., Vulin A., Fougerousse F., Leturcq F., Kaplan J. C., Garcia L., Danos O. Science. 2004;306:1796–1799. [PubMed]
18. Michienzi A., Conti L., Varano B., Prislei S., Gessani S., Bozzoni I. Hum. Gene Ther. 1998;9:621–628. [PubMed]
19. Auricchio A., Kobinger G., Anand V., Hildinger M., O’Connor E., Maguire A. M., Wilson J. M., Bennett J. Hum. Mol. Genet. 2001;10:3075–3081. [PubMed]
20. Chao H., Liu Y., Rabinowitz J., Li C., Samulski R. J., Walsh C. E. Mol. Ther. 2000;2:619–623. [PubMed]
21. Sampaolesi M., Torrente Y., Innocenzi A., Tonlorenzi R., D’Antona G., Pellegrino M. A., Barresi R., Bresolin N., De Angelis M. G., Campbell K. P., et al. Science. 2003;301:487–492. [PubMed]
22. Torrente Y., Belicchi M., Sampaolesi M., Pisati F., Meregalli M., D’Antona G., Tonlorenzi R., Porretti L., Gavina M., Mamchaoui K., et al. J. Clin. Invest. 2004;114:182–195. [PMC free article] [PubMed]
23. Rivera V. M., Gao G. P., Grant R. L., Schnell M. A., Zoltick P. W., Rozamus L. W., Clackson T., Wilson J. M. Blood. 2005;105:1424–1430. [PubMed]
24. Auricchio A., Hildinger M., O’Connor E., Gao G. P., Wilson J. M. Hum. Gene Ther. 2001;12:71–76. [PubMed]
25. Bottinelli R., Canepari M., Pellegrino M. A., Reggiani C. J. Physiol. (London) 1996;495:573–586. [PMC free article] [PubMed]
26. D’Antona G., Pellegrino M. A., Adami R., Rossi R., Carlizzi C. N., Canepari M., Saltin B., Bottinelli R. J. Physiol. (London) 2003;552:499–511. [PMC free article] [PubMed]
