

Партнерка на США и Канаду по недвижимости, выплаты в крипто
- 30% recurring commission
- Выплаты в USDT
- Вывод каждую неделю
- Комиссия до 5 лет за каждого referral
УДК: 539.19
ВЛИЯНИЕ ГАЛОГЕНЗАМЕЩЕНИЯ НА СТРУКТУРУ И ЭЛЕКТРОННЫЕ СЕКТРЫ МОЛЕКУЛ ФЛУОРОНОВЫХ КРАСИТЕЛЕЙ
, ,
научный руководитель канд. хим. наук
Сибирский федеральный университет
Флуороновые красители представляют собой гомологичный ряд соединений на основе флуоресцеина с постепенным замещением атомов водорода атомами галогенов (бром, хлор, йод) (рисунок 1).
|
флуоресцеин (R1=R2=R3=H), 4’5’-дибромфлуоресцеин (R1=R2= H, R3=Br), эозин (R1=H, R2=R3=Br), эритрозин (R1= H, R2=R3=I), бенгальский розовый (R1= Cl, R2=R3=I) Рисунок 1 – Флуороновые красители |
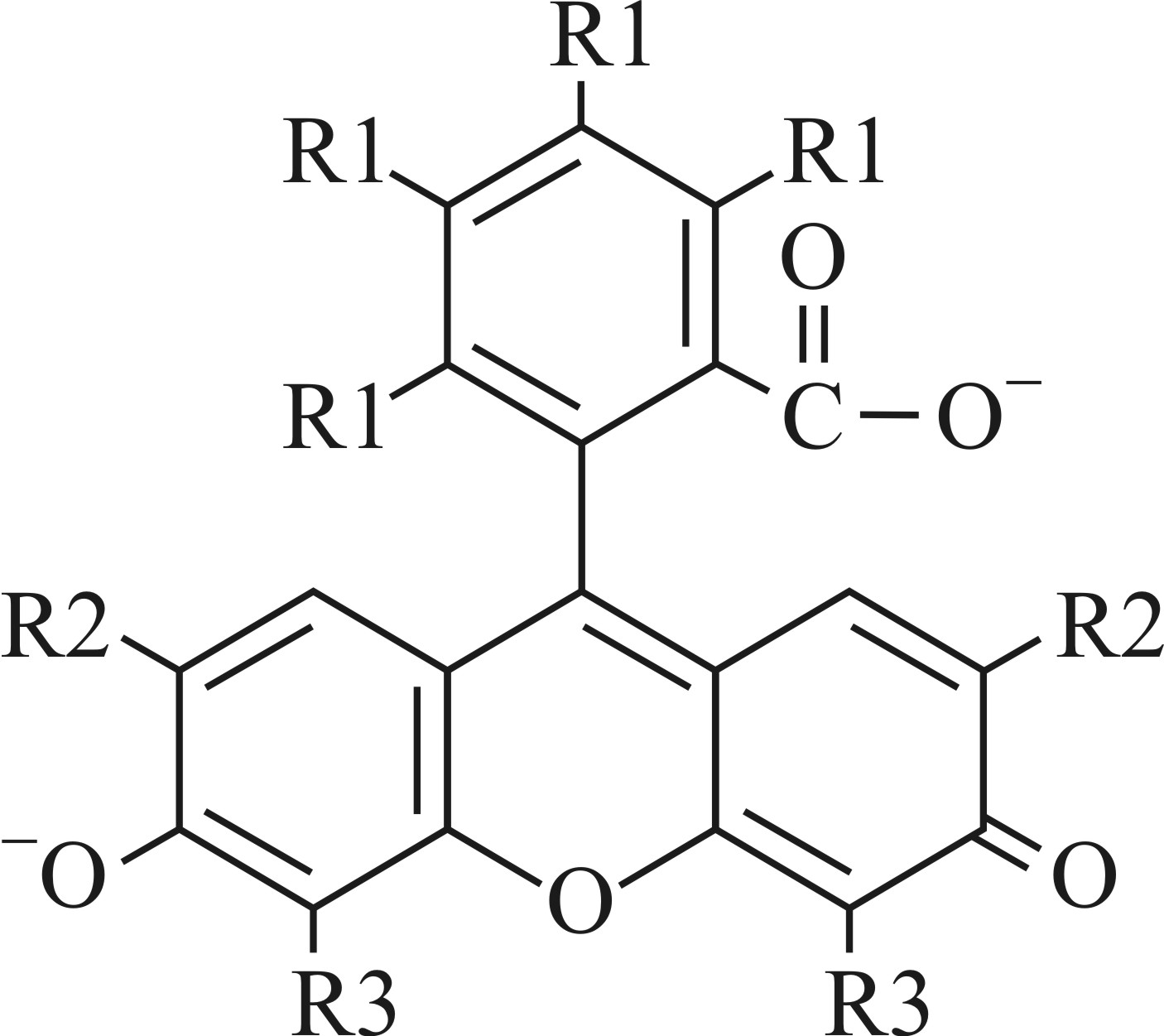
Интерес к исследованию растворов флуороновых красителей обусловлен их применением в качестве активных сред перестраиваемых лазеров, регистрирующих сред для оптической записи информации, флуоресцентных меток для исследования биологических объектов, а также флуоресцентных зондов. Для большинства флуороновых красителей дианионая форма обладает наиболее выраженными абсорбционными и люминесцентными свойствами, которые снижаются у анионной и нейтральной форм. Это позволяет использовать флуороновые красители в качестве ионных индикаторов. Широко известно существенное снижение квантового выхода флуоресценции и увеличение констант скоростей безызлучательных переходов у флуоронов при замещении атомами-галогенами [1]. Галогензамещение вызывает также «красное» смещение полос поглощения флуороновых красителей. Проводилось эмпирическое описание «красного» сдвига длинноволновой полосы поглощения при постепенном замещении атомов водорода в моноцикле и трицикле флуоронов атомами галогенов, но не детальное изучение причин «красного» сдвига.
Квантово-химические методы являются более успешными по сравнению с эмпирическими методами исследования для выявленных закономерностей. Анализ электронных состояний и электронных спектров поглощения дианиона и моноаниона флуоресцеина методом теории функционала плотности (DFT) B3PW91/6-311G позволил выявить различие в их геометрической и электронной структуре [2]. С помощью полуэмпирического метода SCF-MO-CI в приближении Паризера-Парра-Попла, определили энергетическую структуру, энергии переходов, силы осцилляторов и поляризацию переходов дианионов уранина и эозина [3]. Большинство работ посвящено флуоресцеину и его производным, не содержащим тяжелых атомов [4, 5], в то время как его галогенпроизводные (дибромфлуоресцеин, эозин, эритрозин, бенгальский розовый) исследованы меньше. В настоящей работе выявлено влияние заместителей-галогенов на пространственную структуру и электронные спектры дианионов флуороновых красителей результатов квантово-химического расчета электронной структуры (зарядового распределения)
Квантово-химические расчеты характеристик дианионов флуоронов проводились с помощью метода функционала плотности B3LYP [6] в базисах 6-311** и 6-31** программы GAMESS [7]. Для учета влияния растворителя (метанола) использовали модель поляризованного континуума (PCM) Томаса [8], в соответствии с которой растворенная молекула расположена в полости, ограниченной пространством атомных сфер молекулы. Для вычисления длин волн переходов в спектре поглощения использовали метод TD-B3LYP [9]. Частичные электронные заряды представлены по Малликену в единицах абсолютной величины заряда электрона e-. Расчеты проводились для синглетных состояний молекул, имеющих заряд -2 e-. Поиск устойчивой конфигурации молекулы, отвечающей минимуму потенциальной поверхности в вакууме и в растворителе, состоял в оптимизации геометрии основного состояния молекулы методами B3LYP/6-311** и PCM//B3LYP/6-311**. Затем рассчитывались энергии переходов и их силы осцилляторов с помощью методов TD-B3LYP/6-311** и PCM//TD-B3LYP/6-311**. Программа GAMESS позволяет найти геометрию молекулы в возбуждённом состояния только в условиях вакуума. Поэтому сначала конфигурация молекулы в возбуждённом состоянии оптимизировалась без учета растворителя. Затем находились энергии переходов флуоронов в растворителе методом PCM//TD-B3LYP/6-31**. Выбор метода был обусловлен высокой результативностью методов функционала плотности (например, PBE0, B3LYP), сравнимой с возможностями самых сложных пост-хартрифоковских приближений (например, метода конфигурационного взаимодействия).
С помощью квантово-химических методов выявлены конформационные изменения структуры молекул флуоронов в ряду флуоресцеин – розовый бенгальский связанные с перераспределением частичных зарядов, вызывающих изменения длин связей и углов между связями.. Найдены значения постоянных дипольных моментов, дипольных моментов переходов, сил осцилляторов и выявлено смещение положения электронных спектров, обусловленное как галогенированием флуоронов («красный» и стоксов сдвиг), так и «включением» межмолекулярного взаимодействия. Уменьшение стоксова сдвига в ряду флуоронов флуоресцеин – розовый бенгальский качественно подтверждается квантово-химическими методами. Вычисленные значения постоянных дипольных моментов в основном и возбужденном электронных состояниях в метаноле являются завышенными по сравнению с экспериментальными, однако факт увеличения дипольного момента при электронном возбуждении подтверждается экспериментальными результатами. Кислотно-основные свойства дианионов флуоронов соответствуют распределению частичных зарядов на атомах кислорода. Квантово-химические методы дают, в целом, правильную, качественную картину электронных состояний флуоронов, и по мере расширения информативных возможностей выбираемых моделей и методов можно будет достичь более адекватного описания экспериментальных данных.
Литература:
Martin, M. Hydrogen bond effects on radiationless electronic transitions in xanthene dyes / M. Martin // Chem. Phys. Lett. – 1975. – Vol. 35. – P. 105-111. Hirano, K. Electronic Structure and Spectra of Organic Dye Anions of Uranine and Eosin Y / K. Hirano // Bull. Chem. Soc. Jpn. – 1983. – Vol. 56. – P. 850-854. Tamulis, A. Quantum mechanical studies of intensity in electronic spectra of fluorescein dianion and monoanion forms / A. Tamulis, J. Tamuliene, M. L. Balevicius, A. Tamulis // Struct. Chem. – 2003. – Vol. 14. – P. 643-648. Fabian, W. M.F. Effects of annulation on absorption and fluorescence characteristics of fluorescein derivatives: A computational study / W. M.F. Fabian, S. Schuppler, O. S. Wolfbeis // J. Chem. Soc., Perkin Trans. – 1996. – Vol. 2. – P. 853-856. Spagnuolo, C. C. Photostability and spectral properties of fluorinated fluoresceins and their biarsenical derivatives: a combined experimental and theoretical study / C. C. Spagnuolo, W. Massad, S. Miskoski, G. Menendes, N. Garsia, E. Jares'Erisman // Photochem. Photobiol. – 2009. – Vol. 85. – P. 1082-1088 Becke, A. D. Density-functional thermochemistry. The role of exact exchange / A. D. Becke // J. Chem. Phys. – 1993. – Vol.98. – P. 5648-5652. Schmidt, M. W. General Atomic and Molecular Electronic Structure System / M. W. Schmidt, K. K. Baldridge, J. A. Boatz, S. T. Elbert, M. S. Gordon, J. H. Jensen, S. Koseki, N. Matsunaga, K. A. Nguyen, S. , T. L. Windus, M. Dupuis, J. A. Montgomery // put. Chem. – 1993. – Vol. 14. – P. 1347-1363. Cossi, M. Ab initio study of solvated molecules: A new implementation of the polarizable continuum model / M. Cossi, V. Barone, R. Cammi, J. Tomasi // Chem. Phys. Lett. – 1996. – V. 255. – P. 327-335. Gross, E. K.U. Time-dependent density functional theory / E. K.U. Gross, W. Kohn // Ad. Quant. Chem. – 1990. – Vol.21. – P. 255-291.