


Партнерка на США и Канаду по недвижимости, выплаты в крипто
- 30% recurring commission
- Выплаты в USDT
- Вывод каждую неделю
- Комиссия до 5 лет за каждого referral
Для определения точки эквивалентности используют свойство иода окрашивать раствор крахмала (1 %-ный ) в синий цвет.
Иодометрическое определение восстановителей, для которых стандартный окислительно-восстановительный потенциал менее 0,54 В, проводится прямым титрованием рабочим раствором иода. Например,
2S2O32– – 2ē = S4O62– E0 = + 0,08 B
SO32– + H2O – 2 ē = SO42– + 2H+ E0 = – 0,93 B
Sn2+ – 2 ē = Sn4+ E0 = – 0,14 В
При невыполнении условий прямого титрования восстановителей проводят обратное титрование, добавляя к определяемому восстановителю раствор иода в избытке, а остаток иода титруют рабочим раствором Na2S2O3 по реакции:
I2 + 2S2O32– = S4O62– + 2I–
Таким образом определяют например аскорбиновую кислоту.
Иодометрическое определение окислителей, для которых стандартный потенциал выше 0,54 В, проводится титрованием по методу замещения. Заместителем является иод, выделяющийся в эквивалентном окислителю количестве после добавления к нему избытка иодида калия. Например в иодометрии важна реакция окисления иодида дихроматом, используемая при стандартизации рабочего раствора Na2S2O3
Cr2O72–+ 14H+ + 6I– = 2Cr3+ + 3I2 + 7H2O.
Методом замещения проводят также определение кислот, согласно реакции:
6H+ + IO3– + 5I– = 3I2 + 3H2O.
Выделившийся иод оттитровывают раствором тиосульфата.
Растворы иода неустойчивы и изменяют свой титр при хранении вследствие: а) летучести иода; б) его способности окислять различные органические вещества, присутствующие в воде; в) окисления I– кислородом воздуха, которое усиливается на свету в кислой среде:
4I– + O2 + 4H+ = 2I2 + 2H2O
Условия иодометрического титрования ограничиваются тем, что в кислой среде неустойчив тиосульфат, а в щелочной (рН > 8,5) протекают реакции диспропорционирования иода.
Лабораторная работа №4
Определение содержания аскорбиновой кислоты
во фруктовых соках
Аскорбиновая кислота или витамин С является углеводом ряда L- глицеринового альдегида, широко распространенным в природе. Его биологическая роль связана со способностью к окислительно - восстановительным превращениям, сопровождающимся переносом атомов водорода к акцепторам. Витамин С не синтезируется в организме человека и является необходимым пищевым фактором; его недостаток приводит к старению и тяжелым заболеваниям (цинга). Наиболее важными источниками аскорбиновой кислоты для человека служат продукты растительного происхождения (овощи и фрукты): перец, салат, капуста, хрен, укроп, ягоды рябины, черной смородины, цитрусовые. Повседневно организм получает витамин С с картофелем, хотя его содержание в картофеле невелико и снижается за время хранения от 25 до 5 мг/100 г. Из не пищевых источников витамином С богаты шиповник, листья черной смородины, отварами которых можно пополнять запасы его в организме. Суточная потребность взрослых в витамине С составляет 50-100, детей – 30-70 мг.
Окислительно-восстановительный потенциал аскорбиновой кислоты сильно зависит от рН раствора: Е0 = 0,4 В, при рН = 2 значение Е0¢ = 0,28 В, при рН=5,8 Е0¢ = 0,11 В. Определение аскорбиновой кислоты проводится обратным иодометрическим титрованием согласно следующим реакциям:
С6Н8О6 + I2 = С6Н6О6 + 2НI
аскорбиновая дегидро-
кислота аскорбиновая
кислота
I2 + 2S2O32– = S4O62– + 2I–
(остаток)
Реагенты:Раствор иода в KI: с(1/2I2) ~ 0,1 моль/л разбавляют в 10 раз; стандартизованный раствор Na2S2O3 с уточненной концентрацией 0,1 М раствора разбавляют в 10 раз; Н2SO4, 6,0 М раствор; 1 % раствор (индикатор).
Разбавление растворов проводят, помещая в мерную колбу (100,0 мл) аликвоты 10,00 мл соответствующего раствора, доводя до метки и тщательно перемешивая.
Ход анализа. В две колбы для титрования помещают, соответственно, 20 мл дистиллированной воды (холостая проба) и 20,00 мл (V) пробы фруктового сока (или отвара). Добавляют по 4 мл 6 М Н2SO4 и по 10,00 мл 0,01 н. раствора I2. Колбы прикрывают стеклянной пробкой или кусочками стекла и оставляют в темноте на 3 – 5 минут. Затем последовательно титруют рабочим раствором Na2S2O3 с установленной концентрацией до исчезновения синей окраски иод-крахмального комплекса от последней капли титранта, отмечая по бюретке его расход (мл): V1 – на холостую пробу и V2 – на пробу сока. Крахмал добавляют в конце титрования, когда раствор иода приобретет соломенно-желтую окраску. В указанных условиях другие восстановители, например, глюкоза, не реагируют с иодом. Рассчитывают массу (мг) аскорбиновой кислоты (М = 176,1 г/моль) в 100 мл сока:
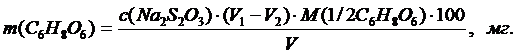
Примечание: для соков с содержанием 5 – 8 мг витамина С на 100 г (по указателю пищевой ценности) объем иода может быть уменьшен до 5,00 мл.
Полученные в работе результаты сопоставляют с характеристиками на упаковках и делают выводы о соответствии сока нормам качества.
1.5. Обработка результатов анализа
При получении результатов анализа, как правило, проводят несколько аналитических операций, используют несколько последовательно измеренных величин, начиная с отбора и подготовки проб. Каждое измерение, каждая операция вносят свой вклад в общую погрешность результата анализа. Операции титриметрического анализа также выполняются с некоторыми, сравнительно небольшими, ошибками. Всякое титриметрическое определение включает в себя: 1) ошибку определения титра рабочего раствора, которая зависит от точности взвешивания и правильности измерения объема: 2) ошибку титрования анализируемого вещества, зависящую от правильности измерения объемов и правильности установления точки эквивалентности с помощью индикаторов (индикаторные сшибки титрования). При выполнении титриметрических определений стремятся к погрешности 0,1 %. Для этого необходимо понимать происхождение погрешностей и уметь их оценивать. Например, для уменьшения случайных погрешностей титрование повторяют несколько раз и берут среднее.
При выполнении анализов контрольных проб (или стандартных образцов), для которых известно истинное содержание (μ) определяемого компонента, вычисляют абсолютную (Δx) и относительную погрешность (Δx отн,%). Если среднее арифметическое значение для n полученных результатов ![]() (
(![]() = (x1 +x2 +х3 + ... + хn)/п), то:
= (x1 +x2 +х3 + ... + хn)/п), то:
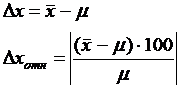
Погрешности классифицируют по характеру причин, их вызывающих. Погрешность определения, обусловленная постоянно действующей причиной, неизменная во всех измерениях (например, сохраняется знак от опыта к опыту) или закономерно изменяющаяся, называется систематической погрешностью. Погрешность, случайным образом изменяющаяся от опыта к опыту, называется случайной погрешностью. Грубые погрешности или промахи резко искажают результат анализа, вызываются небрежностью и, обычно, легко обнаруживаются.
С систематическими и случайными погрешностями связаны, соответственно, правильность и воспроизводимость. Воспроизводимость характеризует рассеяние единичных результатов относительно среднего. Правильность - характеризует отклонение полученного результата от истинного, отражает близость к нулю систематической погрешности. Систематические погрешности выявляются и устраняются или оцениваются и учитываются. Для их выявления используют различные приемы и методы, например, "введено - найдено", анализ стандартного образца, "двойной или тройной добавки".
Оценка случайных погрешностей проводится методами математической статистики. Для ограниченного числа параллельных измерений п (п < 20 – выборочная совокупность данных, выборка) при математической обработке результатов используют распределение Стьюдента, связывающее вероятность попадания величины в данный доверительный интервал, и объем выборки. При этом среднее для ряда параллельных определений 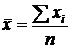 является наиболее вероятным значением измеряемой величины.
является наиболее вероятным значением измеряемой величины.
Характеристиками случайной погрешности (воспроизводимости) являются: выборочная дисперсия S2, стандартное отклонение S и относительное стандартное отклонение Sr.
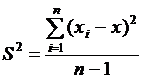 ;
; 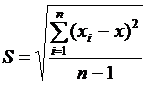 ;
; 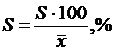
При обработке данных химического анализа определяют границы доверительного интервала (![]() - μ), в котором при заданной доверительной вероятности Р и числе степеней свободы f (f = n-1) лежит истинное значение определяемой величины:
- μ), в котором при заданной доверительной вероятности Р и числе степеней свободы f (f = n-1) лежит истинное значение определяемой величины:
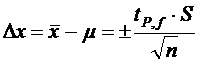 .
.
Значение доверительной вероятности в химическом анализе принято 95 % или 0,95. Это означает, что в рассчитанный интервал попадут 95 из 100 значений. Коэффициент tP,f – коэффициент нормированных отклонений Стьюдента, приведен в справочнике [1] при данных P и f.
С возрастанием числа степеней свободы, т. е. числа параллельных определений значение коэффициента Стьюдента уменьшается, а, следовательно, возрастает точность анализа, поскольку доверительный интервал характеризует воспроизводимость, и в какой то мере, правильность анализа. С учетом доверительного интервала истинное значение полученного результата представляют в виде уравнения:
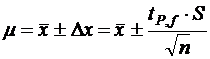 .
.
Оценка промахов (выбраковка результатов). Перед обработкой данных методами математической статистики необходимо выявить промахи и исключить их из числа обрабатываемых результатов. Для выявления промахов используют различные критерии. Самый простой способ выявления промахов – по Q-критерию, который осуществляется следующим образом. Все параллельные результаты располагают в последовательности их убывания или возрастания; затем рассчитывают
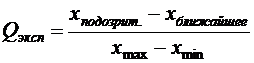
и сравнивают с табличным Qкрит. Если Qэксп < Qкрит., то промах отсутствует и подозрительный результат оставляют в составе выборки. Если же Qэксп > Qкрит, то подозрительное значение является промахом, грубой погрешностью; его отбрасывают. Q – критерий применим к выборкам с n > 5. При малой выборке (п = 3– 5) подозрительный, заметно отличающийся от других, результат просто отбрасывают, а определение повторяют и после этого оценивают случайную погрешность.
Таблица 3.
Значения Q - критерия (при Р = 0,95)
N | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 |
Q | 0,94 | 0,76 | 0,64 | 0,56 | 0,51 | 0,47 | 0,44 | 0,41 |
Пример. При определении содержания аскорбиновой кислоты в пробе картофеля по новой методике пробоподготовки получены следующие результаты (мг/100 г) 14,50; 14,43; 14,54; 14,45; 14,44; 14,52; 14,58; 14,40; 14,25; 14,49. Значение, полученное для той же пробы по стандартной методике, 14,58. Оценить наличие грубых погрешностей, рассчитать среднее и доверительный интервал. Указывают ли полученные результаты на наличие систематической погрешности при работе по новой методике?
1.Наличие промахов оценим по Q–критерию. Располагаем экспериментальные данные в порядке возрастания: 14,25; 14,40; 14,43; 14,44; 14,45; 14,49; 14,50; 14,52; 14,54, 14,58. Проверяем подозрительные значения 14,25 и 14,58. Вычисляем Q –критерий для этих величин:
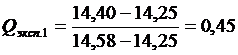 ;
; 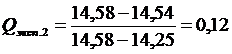 .
.
При Р =0,95 и п = 10 табличное значение Qкрum = 0,41; Qэксп.1 > 0,41, а Qэксп.2 < 0,41, следовательно, значение концентрации 14,25 исключаем, считая недостоверным.
2.Рассчитываем среднее значение концентрации кислоты для выборки с п = 9:
3.Находим стандартное отклонение:
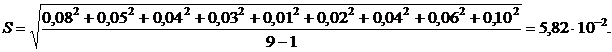
4.Находим границы доверительного интервала, принимая tP, f=2,31 (при Р=0,95 и f=8):
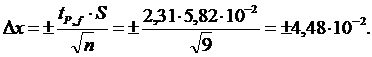
Таким образом, среднее содержание аскорбиновой кислоты лежит в границах
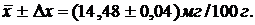
5. Проверим наличие систематической погрешности: истинное значение 14,58 не попадает в доверительный интервал, следовательно, такой метод пробоподготовки картофеля к анализу имеет систематическую погрешность, причину которой надо выяснять.
Правила вычисления. Условие значимости цифр. Результаты анализа должны быть вычислены с той же точностью, что и выполненные измерения. С этой целью при расчете результатов всегда сохраняют одну лишнюю цифру по сравнению с числом цифр в конечном результате, т. е. используют значащие цифры. В конечном результате число округляют и последнюю лишнюю цифру отбрасывают. При определении числа значащих цифр следует помнить, что нули в начале числа незначимы (в числе 0,0015 – две значащие цифры, в 0,0150 – три); нули, стоящие между цифр – всегда значимы. При представлении чисел с нулями, стоящими после цифр, значащие цифры должны быть четко показаны, например, степенным выражением (5,00∙102 – три значащих цифры) или указанием нуля после запятой (200,0 – четыре значащих цифры)
Результат измерения и погрешность следует выражать числом с одинаковым количеством цифр после запятой (см. пример).
Число значащих цифр в результате вычисления произведения или частного величин, измеренных с разной точностью, определяется наименее точным числом
Контрольные вопросы:
(Метод редоксиметрии)
1. Дайте краткую характеристику метода окислительно-восстановительного титрования.
2. Какие способы фиксирования точки эквивалентности применяют в методе окислительно-восстановительного титрования? Что такое редокс-индикаторы?
3. Вычислите молярные массы эквивалентов перманганата калия при его восстановлении в кислой, нейтральной и щелочной среде.
4. Какие приемы титрования применяются в методе окислительно-восстановительного титрования? Приведите примеры.
5. Назовите первичные стандарты, применяемые для установления концентрации перманганата калия.
6. Какими химическими реакциями, протекающими в растворе, обуславливается изменение концентрации перманганата калия?
7. Напишите схему взаимодействия перманганат-иона с оксалат-ионом.
8. Какие восстановители применяют для предварительного восстановления железа(III)? Напишите реакции.
9. Объясните причины погрешностей при восстановлении Fe(III) хлоридом олова (II).
10. Назовите компоненты смеси Рейнгарда-Циммермана и объясните их роль в процессе титрования железа (II).
11. Покажите возможности перманганатометрии в определении разных веществ и их расширение путем выбора приемов титрования?
12. Что называется индуцированной реакцией? Что такое актор, индуктор и акцептор? Чем индуктор отличается от катализатора?
13. Чем отличаются цепные индуцированные реакции от сопряженных индуцированных реакций? Приведите примеры сопряженных реакций.
14. Назовите в системе перманганат – железо (II) – хлорид-ион: а) первичную реакцию; б) индуцированную реакцию; в) актор; г) индуктор; д) акцептор.
15. Перечислите требования, которыми руководствуются при проведении предварительного окисления или восстановления.
16. Охарактеризуйте окислительно-восстановительную способность пары I2/2I–.
17. Назовите первичные стандарты, применяемые для установления концентрации тиосульфата натрия, и приведите уравнения реакций.
18. Назовите индикаторы, применяемые в иодометрии и условия их применения. Почему крахмал прибавляют в самом конце титрования?
19. Как получить чистый иод? Как приготовить раствор иода? Как установить концентрацию раствора иода? Почему раствор иода хранят в сосуде из темного стекла?
20.Как изменяется концентрация тиосульфата натрия во времени? Напишите реакции. Зачем при приготовлении раствора тиосульфата натрия прибавляют карбонат натрия?
21.В связи с чем ограничена возможность проведения реакции тиосульфата с иодом в кислой и щелочной средах?
22.Опишите прямые и косвенные методы в иодометрии и дайте обоснование выбора того или иного метода.
23. Какой прием и способ титрования применяют при иодометрическом определении хлора в воде?
24. Какие формы хлора могут присутствовать в воде при хлорировании? С какой целью их определяют?
25. Как влияет концентрация серной кислоты на иодометрическое определение аскорбиновой кислоты?
26. Предложите схему (условия, уравнения реакций) определения серной и щавелевой кислот при совместном присутствии.
1.6. СОДЕРЖАНИЕ КОЛЛОКВИУМА 2
«Титриметрические методы количественного анализа»
1. Сущность и характеристика титриметрических методов анализа. Техника проведения титриметрического анализа. Мерная посуда и ее характеристики.
2. Классификация по типу реакций (равновесий). Приемы и способы титрования. Примеры прямого, обратного титрования и титрования заместителя. Требования к реакциям в титриметрии. Особенности требований в окислительно-восстановительном титрованиии.
3. Задачи и принципы построения кривых титрования в разных методах титриметрии. Монологарифмические кривые. Понятие скачка титрования. Крутизна кривой и ее значение при выборе условий титрования. Способы установления точки эквивалентности.
4. Способы приготовления рабочих растворов в разных методах титриметрии. Первичные и вторичные стандартные растворы (рабочие растворы с приготовленным и установленным титром).
5. Способы выражения концентрации рабочих растворов. Пересчет концентраций.
6. Расчеты в титриметрии. Вычисление молярной массы эквивалента веществ с учетом протекающих химических реакций. Закон эквивалентов и его использование в расчетах при прямом, обратном титровании и титровании заместителя, способом отдельных навесок и пипетирования. Примеры.
7. Кислотно-основное титрование (метод нейтрализации). Рабочие растворы, исходные вещества. Возможности метода кислотно-основного титрования в анализе различных объектов.
8. Задачи и возможности неводного кислотно-основного титрования.
9. Индикаторы в методе нейтрализации, ионно-хромофорная теория индикаторов. Интервал перехода индикаторов. Выбор индикатора.
10. Происхождение ошибок при титровании с индикаторами. Виды индикаторных ошибок титрования. Примеры.
11. Окислительно-восстановительное титрование (редоксиметрия). Классификация методов по типу титрантов. Характеристика рабочих растворов, исходных веществ.
12. Индикаторы в редоксиметрии, интервал перехода редокс-индикаторов. Специфические индикаторы. Вычисление индикаторной погрешности титрования в редоксиметрии.
13. Приемы титрования при определении неорганических окислителей, восстановителей, органических веществ в перманганатометрии, иодометрии, дихроматометрии, броматометрии. Примеры косвенных определений методами редоксиметрии веществ, не участвующих в окислительно-восстановительных процессах. Предварительное окисление-восстановление определяемых веществ.
14. Факторы, влияющие на скорость окислительно-восстановительных реакций титрования. Значение катализаторов. Каталитические и индуцированные реакции. Индуцированные сопряженные реакции.
15. Выбор кислотности среды в методах перманганатометрии, иодометрии, дихроматометрии, броматометрии.
16. Титрование по методу комплексообразования. Рабочие растворы, классификация методов. Комплексонометрическое титрование аминополикарбоновыми кислотами. Преимущества полидентатных лигандов. Хелатный эффект.
17. Рабочий раствор ЭДТА (трилона Б, комплексона III), условия проведения реакций титрования. Металлохромные индикаторы.
18. Приемы титрования в комплексонометрии. Особенности определения катионов в прямом, обратном, вытеснительном титровании. Возможности определения анионов в косвенном титровании. Маскирующие реагенты. Примеры определений.
19. Сущность метода осадительного титрования. Требования к реакциям в методе осаждения. Классификация методов.
20. Методы аргентометрии. Сравнительная характеристика методов аргентометрии. Рабочие растворы. Индикаторы. Возможности методов Мора, Фольгарда (роданометрии), Фаянса. Методы безиндикаторного титрования в аргентометрии.
21. Метрологические характеристики методик и результатов анализа.
22. Обработка и представление результатов анализа в титриметрии. Происхождение и виды погрешностей (систематические и случайные погрешности). Способы их уменьшения.
23. Законы распределения ошибок для генеральной совокупности данных, для выборки. Доверительный интервал. Необходимое число параллельных определений.
24. Сравнение результатов определения двумя методами (лабораториями).
25. Погрешность единичного определения. Закон сложения погрешностей для суммы (разности) и произведения (частного).
26. Отбор и подготовка проб к анализу как важные аналитические стадии.
2. Физико-химические методы анализа
2.1. Потенциометрия
Потенциометрический метод анализа основан на измерении электродного потенциала, величина которого обусловлена концентрацией (точнее, активностью) потенциалопределяющего компонента раствора.
Для расчета электродного потенциала (Е, В) служит уравнение Нернста:
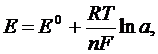
где Е0 – стандартный потенциал, В; R – универсальная газовая постоянная (8,313 Дж); Т – абсолютная температура, К; F – число Фарадея Кл); n – заряд потенциалоопределякщего иона, а – его активность.
После введения численных значений величин R и Т, (температуру принимают равной 298 К (25° С)), и учета коэффициента перехода от натуральных логарифмов к десятичным (2,3026) получают уравнение:
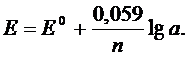
Активность ионов связана с концентрацией с соотношением а = f с, где f – коэффициент активности. В разбавленных растворах коэффициент активности близок к единице, для бесконечно разбавленных растворов уравнение Нернста имеет вид:
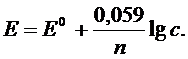
Потенциометрический метод анализа подразделяется на прямую потенциометрию (ионометрия) и потенциометрическое титрование. Прямая потенциометрия основана на измерении потенциала индикаторного электрода и расчете концентрации определяемых ионов по уравнению Нернста. В основе потенциометрического титрования лежит резкое изменение потенциала индикаторного электрода в точке эквивалентности. Используемый в потенциометрических определениях гальванический элемент включает два электрода, которые погружают в один раствор (гальванический элемент без переноса) или в два различных по составу раствора, соединенных жидкостным контактом (цепь с переносом).
Электроды
По назначению электроды делятся на индикаторные и электроды сравнения. Электрод, потенциал которого зависит от активности (концентрации) определяемых ионов в растворе, называют индикаторным. Электрод, потенциал которого не зависит от концентрации определяемых ионов, называется электродом сравнения. В потенциометрическом методе анализа применяют индикаторные электроды двух классов:
1. Электронообменные электроды, на межфазных границах которых протекают реакции, сопровождающиеся переходом электронов.
2. Мембранные или ионоселективные электроды на межфазных границах которых протекают ионообменнные процессы. К таким электродам относится стеклянный электрод, который очень часто используется в потенциометрии.
Устройство комбинированного стеклянного электрода
Электродом является стеклянный шарик (мембрана) диаметром 15-20 мм с толщиной стенок 0,0мм, изготовленный из стекла особого состава (Me2O·Al2O3·SiO2 где Me - Li, Na), расположенный на конце стеклянной трубки (рис.1). Внутри шарика - раствор с определенным значением рН (0,1 ÷ 0,01 M HCI), в который погружен электрод сравнения хлорсеребряный или каломельный.
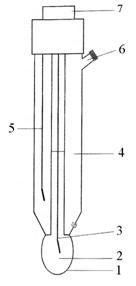
Рис.1. Стеклянный мембранный электрод. 1 – стеклянная мембрана, чувствительная к изменению рН; 2 – внутренний раствор; 3 – внутренний электрод сравнения; 4 – внешний раствор сравнения; 5 – внешний электрод сравнения; 6 – отверстие для заполнения электрода; 7 – электрический контакт.
Перед работой стеклянный электрод некоторое время вымачивается в 0,1 M HCI. При этой ионы H+ из раствора обмениваются на ионы Na+ из мембраны, и в системе устанавливается равновесие:
SiONa+ + H+ ↔ SiOH+ + Na+
стекло раствор стекло раствор
Если подготовленный таким образом электрод опустить в раствор, содержащий ионы Н+, произойдет обмен ионами водорода между анализируемым раствором и внешней поверхностью мембраны, т. е. протекает электродная реакция
H+ ↔ Н+
раствор стекло,
приводящая к возникновению потенциала. Величина этого потенциала зависит от активности ионов Н+ в анализируемом растворе:
![]()
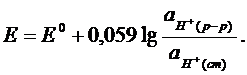
На внутренней поверхности стекла также возникает потенциал, который остается постоянным в растворе с постоянной активностью ионов водорода.
Для определения рН в исследуемый раствор погружается стеклянный индикаторный электрод и хлорсеребряный электрод сравнения (иногда вся система комбинируется в одном электроде – комбинированный электрод).
ЭДС такой ячейки складывается из алгебраической суммы потенциалов, возникающих на отдельных поверхностях разделов фаз; испытуемый раствор - внешняя поверхность стекла (Е1), внутренняя поверхность стекла - стандартный раствор кислоты (Е2), стандартный раствор - вспомогательный хлорсеребряный электрод (E3) и потенциал электрода сравнения, погруженного в исследуемый раствор (Е4):
э. д.с.= Е1 + Е2 + Е3 + Е4 .
Переменным, зависящим от pH исследуемого раствора, является только Е1, поэтому:
э. д.с.= K+ Е1=K+ E0 + 0,059 lg aH+ = K + E0 + 0,059 pH
Стеклянный электрод пригоден для измерения рН от 0 до 10, При рН > 10 возникает "щелочная ошибка" вследствие обмена ионов Na+ из раствора. Для особых сортов стекла, содержащего Сs, La щелочная ошибка мала и измерения можно проводить до рН =14. Кроме того, точность показаний стеклянного электрода снижается в присутствии белков и других органических соединений с большими молекулам, способными адсорбироваться на поверхности стекла.
Правила работы со стеклянным (комбинированным)
электродом
Подготовленный к работе электрод хранят в дистиллированной воде или 0,1 М растворе HCI. Перед проведением измерений электрод следует тщательно промыть дистиллированной водой. Перед погружением в буферный раствор остатки воды удалить осторожным промоканием фильтровальной бумагой.
При погружении электрода в исследуемые растворы следует следить за глубиной погружения: место выхода электрического контакта с внутренним раствором электрода должны находиться в исследуемом растворе. При проведении титрования следует избегать повреждения мембраны якорьком магнитной мешалки, поэтому расстояние от дна стакана для титрования до мембраны должно быть ~1,5 см. После окончания измерения электрод следует промыть и погрузить в стаканчик с дистиллированной водой.
Прибор для потенциометрических измерений.
В данной работе измерение pH проводится некомпенсационным методом на приборе рН-метре "Mera-ELWRO", имеющем высокое входное сопротивление (~1012 Ом). Расположение регуляторов на верхней панели прибора показано на рис.2.
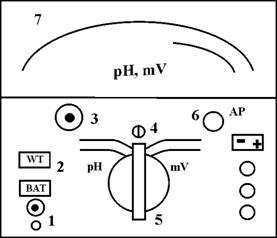
Рис.2. Верхняя панель рН–метра. 1 – клемма для подключения комбинированного электрода; 2 – кнопка включения питания; 3 – регулировка температуры; 4 – механический нуль; 5 – переключатель вида работ; 6 – регулятор настройки по буферным растворам; 7– шкала pH.
Порядок работы на pH-метре
1. Подключить pH–метр к сети 220 B с помощью сетевого шнура.
2. Комбинированный стеклянный электрод подключить pH-метру в положение "1".
3. Положение стрелки прибора должно соответствовать значению шкалы 0. Для установки механического нуля можно воспользоваться "4".
4. Включить прибор нажатием кнопки "2" за 20-30 мин. до проведения измерений.
5. С помощью регулятора ''3" установить нужное значение температуры по шкале температур. Переключатель "5" при этом должен находиться в положении "Т".
6. При проведении измерений рН в интервале 0 – 14 переключатель "5" установить в положение "14 рH", Для точных измерений рН в интервале 0 – 2,8 переключатель установить в положение "2,8 рН".
7. Проверить настройку прибора по двум буферным растворам pH = 4,02 и рH = 9,18. Для этого электрод последовательно погрузить в стаканчик с соответствующим буферным раствором. В случае несовпадения показании по шкале прибора с соответствующим значением рН провести настройку с помощью регулятора "6".
8. После проведения настройки можно приступать к измерению рН исследуемого раствора.
9. После окончания работы прибор выключись, вернув регулятор "5" в положение "Т", отжав кнопку "2"и отключив от сети.
Графические способы установления точки эквивалентности при потенциометрическом титровании.
| Рис.3. Кривые потенциометрического титрования: А – интегральная кривая, Б – дифференциальная кривая. |
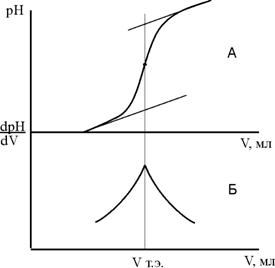
Графические способы определения точки эквивалентности представлены на рис.3.

|
Из за большого объема этот материал размещен на нескольких страницах:
1 2 3 4 |
