

Партнерка на США и Канаду по недвижимости, выплаты в крипто
- 30% recurring commission
- Выплаты в USDT
- Вывод каждую неделю
- Комиссия до 5 лет за каждого referral
На правах рукописи
КОРОЛЕВ Андрей Викторович
ОЦЕНКА ФАРМАЦЕВТИЧЕСКОЙ ЭКВИВАЛЕНТНОСТИ ТВЕРДЫХ ДОЗИРОВАННЫХ ЛЕКАРСТВЕННЫХ ФОРМ С ИСПОЛЬЗОВАНИЕМ ТЕСТА «РАСТВОРЕНИЕ»
15.00.02 — фармацевтическая химия, фармакогнозия
АВТОРЕФЕРАТ
диссертации на соискание ученой степени
кандидата фармацевтических наук
Москва 2009
Работа выполнена в Институте стандартизации и контроля лекарственных средств Федерального государственного учреждения «Научный центр экспертизы средств медицинского применения» (ФГУ НЦЭСМП) Росздравнадзора.
Научный руководитель:
доктор фармацевтических наук,
Официальные оппоненты:
доктор фармацевтических наук, профессор
доктор фармацевтических наук
Ведущая организация:
научный центр по безопасности биологически активных веществ».
Защита состоится «____» ______________ 2009 г. в 14 часов на заседании диссертационного совета Д 208.040.09 при Московской Медицинской Академии имени г. Москва, Никитский бульвар, д. 13
С диссертацией можно ознакомиться в библиотеке ММА имени ( г. Москва, Нахимовский проспект, 49).
Автореферат разослан «____» ______________ 2009 г
Ученый секретарь
диссертационного совета Д.208.040.09
доктор фармацевтических наук,
профессор Наталья Петровна Садчикова
ОБЩАЯ ХАРАКТЕРИСТИКА РАБОТЫ
Актуальность темы. В настоящее время Российский фармацевтический рынок характеризуется большим числом воспроизведенных препаратов (дженериков) как различных зарубежных, так и отечественных производителей. В связи с этим актуальной является проблема изучения и сравнения эффективности воспроизведенных и оригинальных зарубежных и отечественных препаратов.
Эффективность лекарственного препарата (ЛП) системного действия во многом связана с его поступлением в системный кровоток. Для оральных твердых дозированных лекарственных форм (ЛФ) поступление в кровь определенного количества действующего вещества (ДВ) за фиксированный интервал времени определяется следующими этапами: высвобождение ДВ из ЛП, растворение или солюбилизация ДВ в условиях желудочно-кишечного тракта (ЖКТ) и прохождение действующего вещества через стенки ЖКТ.
Смоделировать поведение ДВ in vitro для твердых дозированных форм немедленного высвобождения в большой степени способно испытание «Растворение».
Согласно Руководству ВОЗ по регистрационным требованиям в случае корреляции данных растворения in vitro и абсорбции in vivo, оценка эффективности и безопасности дженериков может проводиться по результатам испытания «Растворение» при сравнении их биоэквивалентности со стандартом, за который принимается оригинальный препарат, выпускаемый предприятием-изготовителем в соответствии с патентным правом на лицензию и эксклюзивную технологию его производства.
С введением в 2003 г. на территории Российской Федерации обязательной фармацевтической экспертизы НД с лабораторной проверкой аналитических методик в ФГУ НЦЭСМП Росздравнадзора испытание «Растворение» проводится в обязательном порядке при процедурах первичной регистрации и перерегистрации в связи с изменением аналитических методик или состава ЛП.
Испытание «Растворение», как известно, также используется при разработке твердых дозированных ЛФ для подбора наиболее подходящих вспомогательных веществ и их пропорций, оптимизации технологического процесса, приведения в соответствие высвобождения ДВ из создаваемых ЛФ с высвобождением из ЛП сравнения, установления стабильности ЛП и подтверждения постоянства качества и характеристик ЛП после внесения изменений в состав ЛФ или производственный процесс (оптимизации производства, смене оборудования или места производства).
Более подробные и объективные сведения об исследуемом ЛП в испытании «Растворение» могут быть получены при изучении высвобождения ДВ не по одной точке, как описано в общей фармакопейной статье «Растворение» и зарубежных фармакопеях, а по профилям растворения с дальнейшим использованием модель-зависимых и модель-независимых методов сравнения полученных профилей растворения, в том числе, коэффициентов подобия и различия, как рекомендовано FDA.
Учитывая вышеизложенное, очевидна актуальность проблемы рационального использования испытания «Растворение» при изучении фармацевтической эквивалентности ЛП на различных этапах их регистрации и перерегистрации, из которой вытекает необходимость разработки унифицированных требований к проведению испытания «Растворение», отвечающих современным возможностям испытания.
Цель и задачи исследования. Целью настоящего исследования является изучение проблемы фармацевтической эквивалентности твердых дозированных ЛФ при внесении изменений в состав уже зарегистрированных ЛП и регистрации воспроизведенных ЛП по результатам теста «Растворение».
Для достижения поставленной цели необходимо было решить следующие основные задачи:
1. Изучить научную литературу и нормативные документы ВОЗ, ЕЭС и FDA по оценке эффективности и безопасности воспроизведённых твердых дозированных ЛФ с использованием испытания «Растворение»; выбрать наиболее подходящий способ сравнения профилей растворения с целью определения фармацевтической эквивалентности твердых дозированных ЛФ.
2. Оценить наличие фармацевтической эквивалентности твердых дозированных ЛФ относительно препаратов сравнения на этапе первичной регистрации или перерегистрации в связи с изменением состава.
3. Сравнить высвобождение воспроизведенных твердых дозированных ЛФ по тесту «Растворение» относительно инновационных ЛП на пострегистрационном этапе (на примере таблеток ципрофлоксацина гидрохлорида и таблеток протионамида различных производителей).
4. Разработать Руководство по проведению испытания «Растворение» с использованием приборов «вращающаяся корзинка» и «лопастная мешалка», включающее в себя рекомендации по выбору типа прибора, среды растворения, общие замечания о проведении испытания «Растворение», а также описание процессов механической калибровки и калибровки с использованием калибраторов USP.
Научная новизна исследования. Проведен сравнительный анализ фармацевтической эквивалентности воспроизведенных ЛП относительно оригинальных ЛП или дженериков с установленной биоэквивалентностью на этапах регистрации, а также ЛП с измененным составом относительно ЛП первоначального состава при перерегистрации с связи с изменением состава на основании изучения профилей растворения 242 пар ЛП отечественных и зарубежных производителей, относящихся к различным фармакотерапевтическим группам.
Показана значимость изучения высвобождения ЛВ из оральных твердых дозированных ЛФ для характеристики качества дженериков по отношению к оригинальным ЛП, а также для контроля за постоянством состава и производственных условий выпускаемых ЛП.
Проведено исследование фармацевтической эквивалентности таблеток, содержащих ципрофлоксацина гидрохлорид и протионамид различных производителей, представленных на отечественном рынке.
Практическая значимость исследования. Результаты исследования фармацевтической эквивалентности использованы при принятии решения о регистрации или перерегистрации испытуемых ЛП.
Разработано Руководство по проведению испытания «Растворение» с использованием приборов «вращающаяся корзинка» и «лопастная мешалка», в которое вошли методические рекомендации по:
- проведению испытания «Растворение»,
- калибровке приборов растворения.
Апробация работы. Материалы диссертации представлены на XII Российском национальном конгрессе «Человек и лекарство» (Москва, 2005), на II всероссийском съезде фармацевтических работников (Сочи, 2005), на заседании Секции №2 Ученого совета ФГУ «НЦ ЭСМП» Росздравнадзора (Москва, 2008).
Публикации. По теме диссертации опубликовано 6 печатных работ.
Связь задач исследования с проблемным планом фармацевтических наук. Диссертационная работа выполнена в соответствии с планом научно-исследовательских работ Института стандартизации и контроля лекарственных средств по теме «Разработка и совершенствование нормативной базы по стандартизации лекарственных средств».
На защиту выносятся:
1. Результаты сравнительного анализа фармацевтической эквивалентности 242 пар ЛП на этапе первичной регистрации и перерегистрации в связи с изменением состава.
2. Результаты изучения фармацевтической эквивалентности на пострегистрационном этапе двух групп препаратов, относящихся к различным классам по системе биофармацевтической классификации.
3. Результаты исследований по калибровке прибора для определения растворения.
Структура и объем диссертации. Диссертация изложена на 168 страницах машинописного текста, состоит из введения, обзора литературы, 3 глав экспериментальной части, общих выводов, списка литературы и 8 приложений. Диссертационная работа проиллюстрирована 46 таблицами и 44 рисунками. Библиографический указатель включает 126 источников, из них 103 на иностранных языках.
ОСНОВНОЕ СОДЕРЖАНИЕ РАБОТЫ
1 калибровка прибора для определения растворения
Для решения поставленных задач использовали результаты исследований, полученных на приборе для растворения Distek 2100C (с автоматическим пробоотборником Agilent на основе перистальтического насоса).
В настоящее время в Государственной фармакопее РФ не описаны приемы и не приведены методики калибровки такого типа оборудования, поэтому первоначально прибор был подвергнут полной механической калибровке (на основании руководства по его эксплуатации), а затем калибровке с использованием калибраторов USP (все остальное необходимое оборудование прошло поверку на соответствие требованиям Госстандарта РФ).
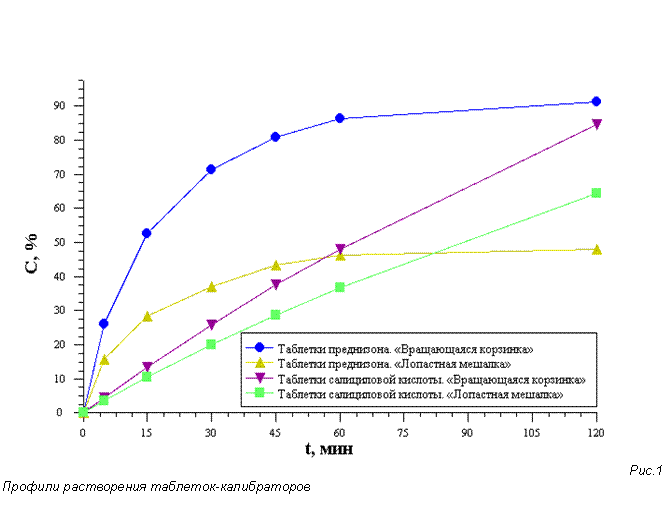
Для калибровки прибора использовали таблетки-калибраторы преднизона и кислоты салициловой (распадающегося и нераспадающегося типов, соответственно, согласно USP). Каждый тип калибраторов исследовали как с использованием «вращающейся корзинки», так и с «лопастной мешалкой» в соответствии с методиками и нормами, приведенными в сертификатах USP на каждую конкретную серию таблеток кислоты салициловой (Salicylic Acid Tablets (Dissolution Calibrator, Non-disintegrating, 30 tablets) и таблеток преднизона (Prednisone Tablets, 30 tablets). Все испытания проводились на 6 таблетках.
Таблица 1
Результаты калибровки прибора Distek 2100C
Тип прибора | Характеристика | Таблетки преднизона | Таблетки кислоты салициловой | ||
Фактически | Норма | Фактически | Норма | ||
Вращающаяся корзинка | Высвобождение (за 30 мин) | 63,9-74,5 % (в среднем — 71,2 %) | 53-77 % | 24,4-26,2 % (в среднем — 25,4 %) за 30 мин | 23-29 % |
RSD | не более 4,3 % для всех точек | не более 20 % для точек ранее 15 мин, не более 10 % для последующих | не более 1,5 % для всех точек | не более 20 % для точек ранее 15 мин, не более 10 % для последующих | |
Лопастная мешалка | Высвобождение (за 30 мин) | 32,7-46,9 % (в среднем — 36,8 %) | 27-48 % | 18,7-20,6 % (в среднем — 19,7 %) | 17-26 % |
RSD | не более 6,8 % для всех точек | не более 20 % для точек ранее 15 мин, не более 10 % для последующих | не более 2,5 % для всех точек | не более 20 % для точек ранее 15 мин, не более 10 % для последующих |
1) Таблетки преднизона (USP), распадающийся тип. Объем среды растворения — 500 мл дегазированной воды; скорость вращения - 50 об/мин (для обоих приборов); продолжительность анализа (время растворения) — 120 мин. Оптическую плотность растворов измеряли на УФ-спектрофотометре в максимуме поглощения при длине волны 242 нм. В качестве стандарта использовали раствор преднизона (USP Reference standard Prednisone, LOT 55900L) с концентрацией около 0,02 мг/мл.
2) Таблетки кислоты салициловой (USP), нераспадающийся тип. Объем среды растворения — 900 мл дегазированного 0,05 М фосфатного буфера с рН 7,40 ± 0,05; скорость вращения — 100 об/мин (для обоих приборов); продолжительность анализа (время растворения) — 120 мин. Оптическую плотность растворов измеряли на УФ-спектрофотометре в максимуме поглощения при длине волны 296 нм. В качестве стандарта использовали раствор кислоты салициловой (USP Reference standard Salicylic Acid, Lot J2B147) с концентрацией около 0,30 мг/мл.
Из результатов, представленых в таблице 1, следует, что испытуемый прибор удовлетворяет требованиям, предъявляемым USP к калибровке на таблетках-калибраторах, как по уровню высвобождения ДВ, так и по относительному стандартному отклонению (RSD). Профили растворения таблеток-калибраторов представлены на рис. 1. В дальнейшем калибровка прибора проводилась согласно установленным нормам — раз в полгода.
2 Оценка фармацевтической эквивалентности ЛЕКАРСТВЕННЫХ ПРЕПАРАТОВ на этапе регистрации
В рамках поставленных задач проводили изучение профилей растворения индивидуальных ЛП и сравнительную оценку эквивалентности двух ЛП (всего 242 пары), которая была использована в статистическом анализе результатов, полученных в испытаниях «кинетика растворения». Испытания проводили по методикам, предусмотренным нормативными документами на ЛП сравнения. Предварительно изучаемые ЛП проверялись на соответствие требованиям действующих нормативных документов.
Расчет содержания действующих веществ (ДВ) в среде растворения проводили по общепринятым формулам, включенным в рассматриваемые НД.
Содержание ДВ в среде растворения в каждый интервал времени рассчитывали по формуле:
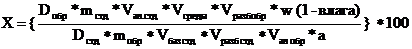
где Dобр - оптическая плотность раствора образца препарата
Dстд - оптическая плотность раствора стандартного образца
mстд - масса навески стандартного образца, мг
a - номинальное содержание действующего вещества в препарате, мг
Vбаз стд - объем базового (первого) разведения раствора стандартного образца, мл
Vал. стд - объем аликвоты, взятой для второго разведения раствора стандартного образца, мл
Vразб стд - объем второго разведения раствора стандартного образца, мл
Vсреды - объем среды растворения, мл
Vал обр - объем аликвоты среды растворения, взятой для разведения, мл
Vразб обр - объем, до которого разводилась среда растворения, мл
w - количественное содержание («чистота») стандартного образца, в долях единицы
влага - содержание влаги в препарате, в долях единицы
100 — коэффициент пересчета результата в проценты
При проведении анализа по нескольким точкам использовали следующие методики:
· после отбора аликвоты (фиксированного объема) среда растворения в стакане пополняется свежей порцией среды растворения, равной объему отобранной аликвоты. В этом случае при расчетах производили коррекцию результатов по следующим формулам:

где значения нижних индексов соответствуют порядку отбора пробы, например, Хиспр 1 – концентрация первой пробы,
Хиспр.2 – исправленная концентрация второй пробы и т. д.; концентрации представлены не в процентах, а в частях.
· после отбора аликвоты (фиксированного объема) среда растворения в стакане не пополняется. В данном случае отбираемый объем аликвоты учитывали при вычислении концентраций ДВ во второй и последующей точках; т. е. в качестве объема среды растворения в каждой точке отбора, начиная со второй, использовали объем, равный объему в предыдущей точке отбора за вычетом объема аликвоты:
![]()
где Vn — объем среды растворения в n-ой точке отбора, мл
Vнач — объем среды растворения в начале испытания, мл
Vал — объем аликвоты, мл
Сопоставимость полученных профилей растворения в испытаниях оценивали с помощью коэффициента различия и коэффициента подобия, принятых Center for Drug Evaluation and Research (CDER) и Human Medicines Evaluation Unit of The European Agency for the Evaluation of Medicinal Products (EMEA) в качестве критериев оценки подобия профилей растворения "in vitro".
Коэффициент различия (ƒ1):
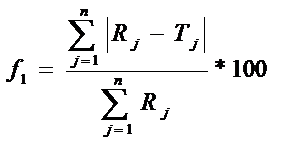
Коэффициент подобия (ƒ2):
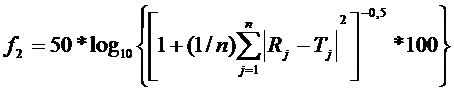
где: n – число пробоотборов,
Rj и Tj – процент растворившегося вещества из стандартного и испытуемого ЛП в каждый момент времени j.
Профили растворения, как известно, являются подобными, если значение f1 находится в пределах диапазона от 0 до 15 и значение f2 находится в пределах диапазона от 50 до 100.
Однако, вычисление коэффициентов различия и подобия представлялось возможным лишь для тех случаев, когда профили растворения ЛП содержат не менее 3 точек, из которых допускается наличие не более одной точки после высвобождения 85 % ДВ или после выхода на плато, а также относительное стандартное отклонение (CV, RSD) должно быть < 20 % для первых точек и < 10 % для остальных или же средняя разница между образцом и стандартом в каждой точке должна быть < 15 %. В тех случаях, когда в сравниваемых ЛФ высвобождалось 85 % и более ДВ за 15 минут, согласно рекомендациям FDA, они считались эквивалентными.
В таблице 2 представлено распределение испытуемых ЛП различных фармакотерапевтических групп по их международному непатентованному наименованию (91 МНН); 13 пар ЛП не имели МНН.
Сравнительные исследования профилей растворения позволили оценить эквивалентность испытуемого ЛП относительно стандарта, в качестве которого выступал аналогичный ЛП другого производипары) или сравнить эквивалентность ЛП одного и того же производителя после внесения изменений в состав вспомогательных веществ ЛФ (88 пар). В данном случае в качестве препарата сравнения выступал ЛП первоначального состава.
Результаты исследований 3-х ЛП не были отнесены ни к одной из двух выше представленных групп. Для ЛП [Andrew Ko1] «кальмабен, таблетки, покрытые оболочкой, 50 мг», производства «Фармацевтический завод Монтавит ГмбХ», Австрия, было признано нецелесообразным сравнение с таблетками димедрола. В двух случаях для таблеток калия иодида отсутствовал показатель «Растворение».
Таблица 2
Распределение испытуемых ЛС по МНН
№ п/п | МНН | Число пар ЛП | № п/п | МНН | Число пар ЛП | № п/п | МНН | Число пар ЛП |
1 | азитромизин | 4 | 32 | индапамид | 4 | 63 | пипемидиновая кислота | 2 |
2 | амисульприд | 1 | 33 | итраконазол | 2 | 64 | пирацетам | 2 |
3 | амлодипин | 9 | 34 | калия иодид | 2 | 65 | преднизолон | 2 |
4 | амоксициллин | 1 | 35 | калия оротат | 3 | 66 | пропафенон | 1 |
5 | ампициллин | 1 | 36 | каптоприл | 1 | 67 | рамиприл | 2 |
6 | анаприлин | 3 | 37 | карбамазепин | 1 | 68 | ранитидин | 3 |
7 | аспарагинат калия и магния | 1 | 38 | карведилол | 13 | 69 | рибавирин | 1 |
8 | атенолол | 8 | 39 | кетотифен | 1 | 70 | рифампицин | 1 |
9 | аторвастатин | 4 | 40 | кларитромицин | 5 | 71 | рокситромицин | 5 |
10 | ацетилсалициловая кислота | 2 | 41 | клиндамицин | 1 | 72 | сертралин | 1 |
11 | ацикловир | 2 | 42 | левотироксин | 1 | 73 | симвастатин | 11 |
12 | ацитретин | 2 | 43 | левофлоксацин | 2 | 74 | тамоксифен | 2 |
13 | бетагистин | 2 | 44 | лизиноприл | 3 | 75 | тамсулозин | 2 |
14 | бикалутамид | 1 | 45 | лозартан | 1 | 76 | телмисартан | 2 |
15 | вальпроевая кислота | 4 | 46 | ломустин | 1 | 77 | тинидазол | 2 |
16 | венлафаксин | 1 | 47 | лоперамид | 1 | 78 | тиоридазин | 2 |
17 | верапамил | 4 | 48 | лоратадин | 2 | 79 | трамадол | 2 |
18 | винпоцетин | 1 | 49 | меалазин | 1 | 80 | триметазидин | 3 |
19 | габапентин | 3 | 50 | мебендазол | 2 | 81 | фамотидин | 3 |
20 | гидроксикарбамид | 1 | 51 | мелоксикам | 2 | 82 | фексофенадин | 2 |
21 | гидрохлоротиазид | 1 | 52 | метилурацил | 1 | 83 | флуоксетин | 1 |
22 | глибенкламид | 2 | 53 | метопролол | 1 | 84 | флюконазол | 2 |
23 | гликлазид | 1 | 54 | метронидазол | 2 | 85 | фтивазид | 3 |
24 | глимепирид | 4 | 55 | метформин | 3 | 86 | фурадонин | 1 |
25 | глюкозамин | 1 | 56 | мифепристон | 1 | 87 | цетиризин | 3 |
26 | диклофенак | 4 | 57 | нитразепам | 1 | 88 | цефалексин | 1 |
27 | доксазозин | 1 | 58 | омепразол | 4 | 89 | ципрофлоксацин | 7 |
28 | домперидон | 2 | 59 | орнидазол | 1 | 90 | эналаприл | 5 |
29 | дротаверин | 3 | 60 | офлоксацин | 3 | 91 | этацизин | 1 |
30 | ибупрофен | 5 | 61 | парацетамол | 3 | 92 | отсутствует | 13 |
31 | изониазид | 4 | 62 | пентоксифиллин | 2 |
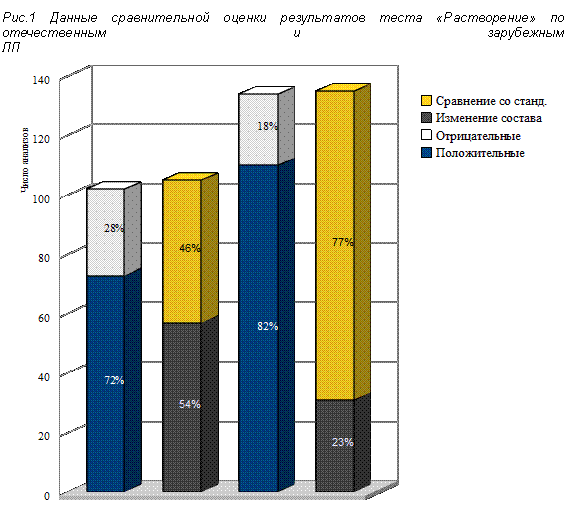 |
Проанализировано 107 отечественных препаратов 27 производителей и 135 препаратов зарубежных производителей из 23 стран.

|
Из за большого объема этот материал размещен на нескольких страницах:
1 2 |
