


Партнерка на США и Канаду по недвижимости, выплаты в крипто
- 30% recurring commission
- Выплаты в USDT
- Вывод каждую неделю
- Комиссия до 5 лет за каждого referral
Аналитический обзор по теме «Алгоритмы сборки геномных последовательностей»
(фамилия, имя, отчество)
В 1958 году Ф. Криком было сформулировано правило, названное впоследствии «Центральной догмой молекулярной биологии». Она постулирует правило передачи информации от ДНК через РНК к белку. В дальнейшем были открыты и иные переходы информации (обратная транскрипция, репликация). Таким образом, было обосновано три принципиально важных уровня хранения и реализации биологической информации: геном, транскиптом и протеом.
Геном представляет собой полный набор генов и иного, некодирующего наследственного материала организма, содержащегося в гаплоидном наборе хромосом и внеядерных элементах. Иными словами, вся генетическая информация организма хранится в геноме. Там же происходят ее изменения.
Транскриптом – это набор всех транскриптов (матричных РНК и некодирующих РНК) клетки. Важно подчеркнуть, что транскриптом в значительной мере подвержен влиянию как внешних так и внутренних факторов и, в отличие от генома, не является постоянным.
Протеом определяет полный набор белков клетки или группы клеток (в общем случае – организма). Так же, как и транскриптом, протеом не является постоянным. Под действием внутренней и внешней среды изменяется как сам набор белков, так и количественные соотношения различных его компонентов. Изменения транскриптома и протеома являются одним из важнейших факторов общего адаптационного синдрома и различных форм иммунного ответа организма.
При изучении генома живого существа обычно выделяют три основных этапа:
а) секвенирование молекул ДНК, содержащих информацию о геноме (выполняется с использованием специальных устройств-секвенаторов);
б) сборка геномной последовательности (или коротко – сборка генома, выполняется с использованием компьютеров);
в) анализ и сравнение геномов (выполняется с использованием компьютеров).
Одним из основных этапов по получению полногеномных последовательностей является биоинформатическая составляющая. К примеру, при использовании геномных анализаторов GAIIx речь идет о составлении первичной последовательности ДНК человека (около 3 млрд нуклеотидов, а с учетом того, что геном каждого человека несет одну свою составляющую от отца, а одну копию от матери, то задача состоит в определении последовательности 6 млрд нуклеотидов) из достаточно коротких (50-100 нуклеотидов) фрагментов, из которых и нужно выстроить гигантскую последовательность длиной 6 млрд нуклеотидов (см. рис. 1). Технический прорыв в создании секвенаторов второго поколения остро поставил вопрос о существовании быстрых алгоритмов биоинформатической сборки генома.
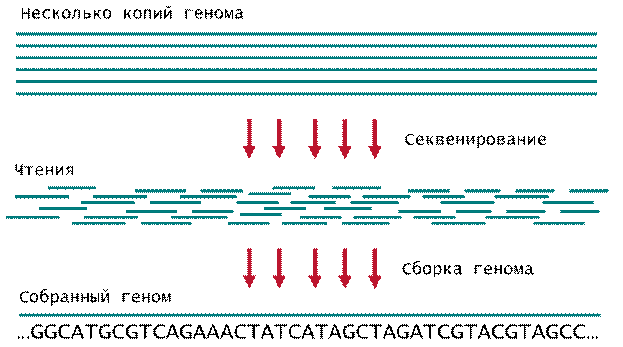
Рис. 1. Секвенирование и сборка генома
Задача разработки методов сборки геномных последовательностей является, в определенном смысле, центральной среди всех задач анализа последовательностей в теоретической и прикладной геномике. Это объясняется тем, что без ее решения нельзя приступить к детальному изучению генома живого существа и его анализу с применением алгоритмов биоинформатики.
В середине первого десятилетия XXI века широкое распространение получили так называемые технологии next generation sequencing (технологии секвенирования нового поколения). По оценкам экспертов [1] эти технологии в настоящее время развиваются быстрее, чем компьютерные технологии и алгоритмы сборки геномных последовательностей – производительность компьютеров удваивается каждые два года, а производительность геномных секвенаторов за тот же самый период увеличивается в 10 раз.
Таким образом, актуальной является задача разработки новых методов и алгоритмов сборки генома, соответствующих по своим параметрам существующим методам секвенирования. Одной из задач, широко решаемой в мире, является задача de novo сборки генома – сборки генома живого существа, для которого геном еще не известен.
Сложность задачи сборки геномной последовательности обусловлена следующими факторами:
г) большой объем входных данных, который составляет десятки и сотни гигабайт;
д) сложность структуры генома – наличие в нем повторов и полиморфизмов;
е) наличие ошибок в исходных данных, полученных с устройств-секвенаторов.
Для того чтобы уменьшить влияние некоторых факторов, вызывающих ошибки, геномную последовательность покрывают чтениями несколько десятков раз. При этом покрытие геномной последовательности чтениям оказывается достаточно равномерным – все позиции, независимо от их расположения в последовательности, покрыты чтениями примерно одинаковое число раз.
В настоящее время исследования в области разработка алгоритмов для решения задач анализа последовательностей, возникающих в теоретической и прикладной геномики, ведутся в таких университетах и лабораториях мира, как, например, Cold Spring Harbor Laboratory (штат Нью-Йорк, США), Университета Мериленда (США), Национальный центр геномного анализа (Барселона, Испания).
Изучение генома человека и других живых существ имеет важное прикладное значение. На основании результатов сборки генома конкретного человека возможна реализация персонифицированной медицины – определения предрасположенности человека к различным болезням, создание индивидуальных лекарств и т. д. Кроме этого, на основе результатов исследования геномов растений и животных с использованием методов биоинженерии могут быть выведены новые их виды, обладающие определенными свойствами.
Еще одно важное приложение исследования ДНК — генетические заболевания. У особей, зараженных одним генетическим заболеванием, наблюдаются одинаковые изменения в ДНК, что может быть использовано в медицине как для теоретического исследования заболевания, так и для лечения от него.
Существует большое число сборщиков генома. Например, ABySS (Assembly ByShort Sequences) [2], Velvet [3], SOAPdenovo [4], Contrail [5] и др.
Проводить сравнение сборщиков с разными подходами очень сложно, как теоритическое, так и практическое. Однако существуют конкурсы, которые направлены на сравнение результатов работы разнообразных сборщиков. Один из них – проект de novo Genome Assembly Project (dnGASP, организован Национальным центром геномного анализа, Барселона, Испания, http://cnag. bsc. es).
В рамках проекта dnGASP участникам требовалось за время с 15 декабря 2010 года по 15 февраля 2011 года (в дальнейшем этот срок был продлен до 1 марта 2011 года) выполнить сборку генома из данных, предоставленных организаторами проекта. Организаторами проекта был подготовлен искусственный геном размером в 1,8 млрд нуклеотидов, из которого были симулированы чтения секвенатора нового поколения. Выбор искусственного генома обосновывается тем, что в таком случае проще проводить сравнение результатов (так как геном известен организаторам) и проще обеспечить честность соревнования (так как геном неизвестен участникам и отсутствует в геномных базах данных).
Большинство из представленных выше сборщиков приняли участие в конкурсе dnGASP.
По результатам этого конкурса стало понятно, что если качество сборки у сборщика является хорошим, то необходимые вычислительные ресурсы на обеспечение такой сборки остаются крайне высокими, что не позволяет использовать данные сборщики в производственном масштабе. Были и сборщики с меньшими требованиями к вычислительным ресурсам, однако качество у таких сборщиков было не самым высоким.
Список литературы
1. Зубов для чтения ДНК // Химия и жизнь. 2010, №7, с. 4 – 7.
2. Simpson J. T., Wong K., Jackman S. D., Schein J. E., Jones S. J. M., Birol I. Abyss: a parallel assembler for short read sequence data. // Genome Res. Jun 2009. Vol. 19, no. 6. Pp. 1117–1123.
3. Zerbino D. R., Birney E. Velvet: algorithms for de novo short read assembly using de Bruijn graphs. // Genome Res. May 2008. Vol. 18, no. 5. Pp. 821–829.
4. Li R., Zhu H., Ruan J., Qian W., Fang X., Shi Z., Li Y., Li S., Shan G., Kristiansen K., Li S., Yang H., Wang J., Wang J. De novo assembly of human genomes with massively parallel short read sequencing // Genome Research. 2010. Vol. 20, no. 2. Pp. 265–272.
5. Schatz M. High throuput sequence analysis with MapReduce. JCVI Informatics Seminar, June 2009. http://www. cbcb. umd. edu/~mschatz/Presentations/.JCVI. pdf
Соискатель _____________________
О.
