

Партнерка на США и Канаду по недвижимости, выплаты в крипто
- 30% recurring commission
- Выплаты в USDT
- Вывод каждую неделю
- Комиссия до 5 лет за каждого referral
![]() - «Копировать атом» копирует строку, если предварительно ее отметить мышью в первой колонке ID.
- «Копировать атом» копирует строку, если предварительно ее отметить мышью в первой колонке ID.
![]() - «Инверсия» изменяет координаты атомов в строке на противоположные по знаку, если предварительно строку отметить мышью в первой колонке ID.
- «Инверсия» изменяет координаты атомов в строке на противоположные по знаку, если предварительно строку отметить мышью в первой колонке ID.
![]() - «Трансформация» открывает окно «Преобразование координат» для строки, отмеченной в первой колонке ID мышью.
- «Трансформация» открывает окно «Преобразование координат» для строки, отмеченной в первой колонке ID мышью.
![]() - «Рассчитать и сохранить данные» отмечает параметры, зафиксированные элементами симметрии, желтым цветом, рассчитывает кратность позиции и сохраняет полученные данные.
- «Рассчитать и сохранить данные» отмечает параметры, зафиксированные элементами симметрии, желтым цветом, рассчитывает кратность позиции и сохраняет полученные данные.
![]() - «Закрыть» закрывает окно «Атомные данные для фазы - *».
- «Закрыть» закрывает окно «Атомные данные для фазы - *».
3.6. Окно «Преобразование координат»
Окно «Преобразование координат» появится после выбора инструмента ![]() - «Трансформация» в окне «Атомные данные для фазы - *».
- «Трансформация» в окне «Атомные данные для фазы - *».

Симметричное преобразование - из числа точек общего положения выбирают номер симметричного преобразования. Все параметры в матрице преобразования заданы жестко. Изменить можно только смещение атомов относительно своих позиций в четвертой колонке s.
Преобразование пользователя - вводят матрицу преобразования и смещение атомов s (четвертая колонка).
Для того чтобы применить преобразование для одного атома, для отмеченных атомов или для всех атомов, следует в окне «Атомные данные для фазы - *» выделить необходимые атомы с помощью клавиши SHIFT.
«OK» - выходит в окно «Атомные данные для фазы - *», где атомные координаты изменяются в соответствии с заданными матрицами преобразования.
«Отмена» - закрывает окно «Атомные данные для фазы - *» без запоминания внесенных данных.
3.7. Данные атомного рассеяния
После выбора кнопки ![]() - «Применить 2» в окне «Структурные данные и параметры пиков» могут возникнуть две проблемы:
- «Применить 2» в окне «Структурные данные и параметры пиков» могут возникнуть две проблемы:
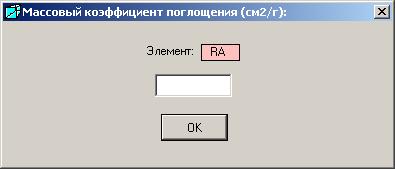
1. В программе нет данных массового коэффициента поглощения для какого-то элемента.
Появится сообщение «Массовый коэффициент поглощения для элемента … не табулирован. Введите коэффициент поглощения». Следует найти его значение в справочной литературе и ввести в окне «Массовый коэффициент поглощения (см2/г)».
2. В программе нет данных для какого-либо излучения.
Появится окно «Данные атомного рассеяния».

Данные атомного рассеяния вводят, нажимая кнопку «Применить», последовательно для каждого элемента, входящего в формулу. После ввода массовых коэффициентов поглощения для всех элементов становится доступной кнопка «OK».
Если коэффициенты аномального рассеяния не известны, то их можно не задавать. По умолчанию они будут иметь нулевые значения.
«OK» - выходит в окно «Структурные данные и параметры пиков».
«Отмена» - закрывает окно без запоминания внесенных данных.
3.8. Ввод параметров пиков для расчета полуширины линий
В окне «Структурные данные и параметры пиков» можно задать разные значения параметров расчета полуширины пиков для разных типов рефлексов, т. к. рефлексы, относящиеся к определенному типу, могут иметь полуширину, отличную от остальных рефлексов.
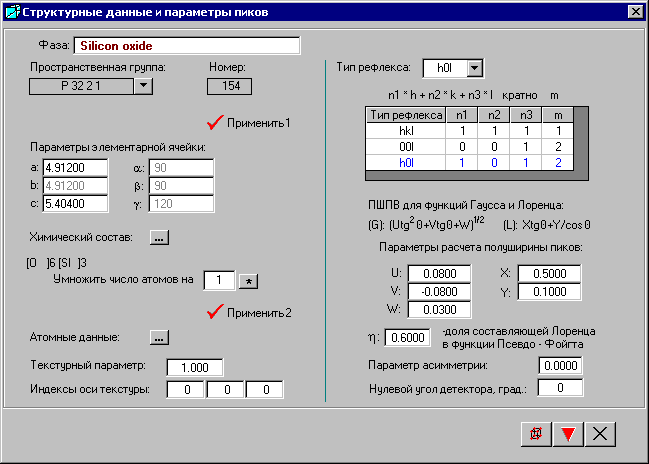
По умолчанию в таблице всегда стоит тип рефлекса hkl. Любой другой тип выбирают из открывающегося списка. Выбранный тип рефлекса заносится в таблицу, обозначается синим цветом и выводит значения параметров, соответствующие этому типу рефлексов. Двойной щелчок мышью на строке таблицы удаляет этот тип рефлекса из таблицы.
Если таблица содержит только одну строку, то указанные параметры расчета полуширины пиков подходят для всех типов рефлексов независимо от того, какой тип рефлекса указан в первой строке таблицы.
Ширина пика на половине его высоты ПШПВ для составляющей функции Гаусса (G) в функции псевдоФойгта рассчитывается по формуле (U g2q + V tgq + W)1/2.
Ширина пика на половине его высоты ПШПВ для составляющей функции Лоренца (L) в функции псевдоФойгта рассчитывается по формуле X tgq + Y/cosq.
Профиль рентгеновских пиков аппроксимируется дублетной функцией псевдоФойгта:
pV = hL + (1- h)G,
где pV - функция псевдоФойгта,
L - функция Лоренца (Коши),
G - функция Гаусса,
h - фактор формы, изменяется от 0 до 1.
Функция Лоренца имеет острую вершину и пологие склоны и хорошо описывает форму отражения, получаемого от образца.
Функция Гаусса имеет пологую вершину и быстро спадающие склоны и хорошо описывает форму отражения, которое дает гониометр.
Долю функции Лоренца в составе функции Псевдо-Фойгта можно определить самим. Например, если h = 0.6, то доля функции Лоренца составит 60%, а доля Гаусса - 40%.
Параметр асимметрии – это отношение левой половины полуширины пика к правой половине. По умолчанию стоит значение 0, т. е. пики для данной фазы симметричные.
Параметр асимметрии рассчитывается по формуле (J. Appl. Cryst., 1995, 28, p. 115-120):
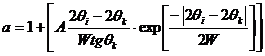 ,
,
где W - ширина пика на половине его высоты ПШПВ,
2qk – угол в максимальной точке пика,
2qi – текущий угол.
4. Окно «Рассчитанные структурные данные»
4.1. Общие данные
Общие рассчитанные данные для фазы можно посмотреть, выбрав кнопку ![]() - «Показать рассчитанные данные» в окне «Структурные данные и параметры пиков».
- «Показать рассчитанные данные» в окне «Структурные данные и параметры пиков».
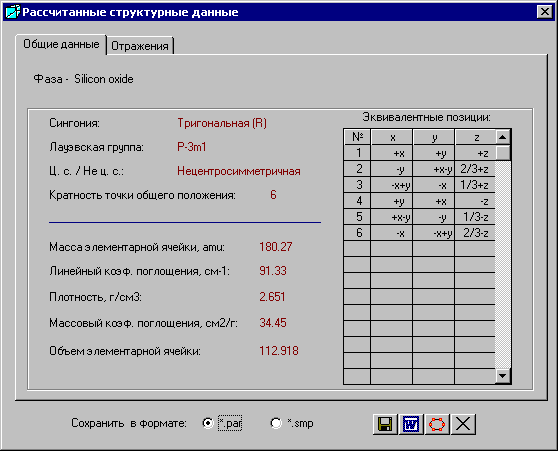
Сингония, лауэвская группа, симметричность, кратность точки общего положения рассчитываются из сингонии. Масса элементарной ячейки, линейный коэффициент поглощения, плотность, массовый коэффициент поглощения рассчитываются из химического состава.
Эквивалентные позиции рассчитываются из атомных данных.
Если выбрана радио-кнопка «Сохранить в формате *.par», то сохраняются структурные данные. Если выбрана радио-кнопка «Сохранить в формате *.smp», то сохраняются отражения в файле, который можно использовать в других программах комлекса PDWin, в частности, в количественном анализе при калибровке по чистым фазам, рассчитанным по структурным данным.
![]() - «Сохранить как» открывает стандартное окно «Сохранить как» для сохранения фазы в файле в форматах *.par и *.smp.
- «Сохранить как» открывает стандартное окно «Сохранить как» для сохранения фазы в файле в форматах *.par и *.smp.
![]() - «Копировать» открывает текстовый редактор MSWord с результатами расчета. Если ранее был открыт документ MSWord, то результаты будут внесены в тот же документ.
- «Копировать» открывает текстовый редактор MSWord с результатами расчета. Если ранее был открыт документ MSWord, то результаты будут внесены в тот же документ.
![]() ВНИМАНИЕ! Если MSWord на компьютере не установлен, то следует открыть редактор WordPad и выбрать кнопку
ВНИМАНИЕ! Если MSWord на компьютере не установлен, то следует открыть редактор WordPad и выбрать кнопку ![]() - «Вставить».
- «Вставить».
![]() - «Геометрия» открывает окно «Геометрия».
- «Геометрия» открывает окно «Геометрия».
![]() - «Закрыть» закрывает данное окно.
- «Закрыть» закрывает данное окно.
4.2. Окно «Сохранить как» - сохранение в форматах *.par и *.smp
Окно «Сохранить как» появится после выбора инструмента ![]() - «Сохранить как» в окне «Рассчитанные структурные данные/Общие данные».
- «Сохранить как» в окне «Рассчитанные структурные данные/Общие данные».
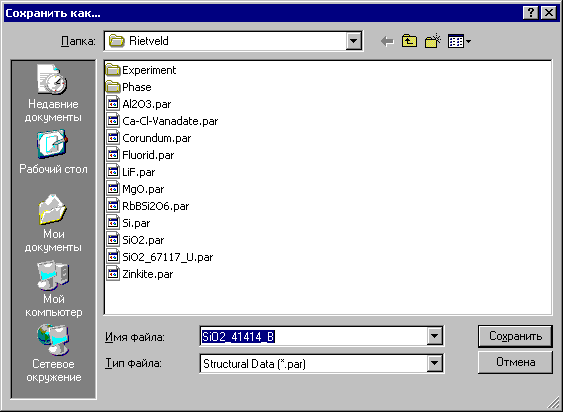
Существуют две возможности сохранить файл фазы под любым именем в любом каталоге (см:
1. StructuralData (*.par) - образуется файл со структурными данными и два дополнительных файла *.hkp – файл с рассчитанными интенсивностями и структурными факторами и *.spr – файл с характеристиками пиков.
2. PDWin files (*.smp) - образуется файл с рассчитанной дифракционной картиной. Файл с таким расширением образуется после работы программы «Предварительная обработка - DRWin» комплекса PDWin и используется многими его программами, в частности, программой «Количественный анализ – Quan», когда нет возможности найти чистые фазы для калибровки. При выборе данной возможности открывается окно «Что хранить»:
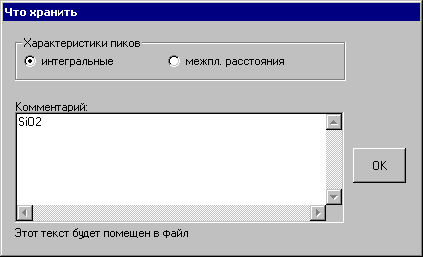
При выборе интегральных характеристик пиков сохраняются углы 2q центров тяжестей отражений и интегральные интенсивности дублетной линии a1+a2.
При выборе межплоскостных расстояний сохраняются относительные интенсивности (приведенные к 100) и межплоскостные расстояния (a2 линии отбрасываются).
![]() ВНИМАНИЕ! Каждый вид информации сохраняется в отдельном файле.
ВНИМАНИЕ! Каждый вид информации сохраняется в отдельном файле.
С клавиатуры набирается любой комментарий, который будет помещен в файл образца.
4.3. Отражения
Закладку «Отражения» выбирают в окне «Рассчитанные структурные данные», которое появится после выбора кнопки ![]() - «Показать рассчитанные данные» в окне «Структурные данные и параметры пиков».
- «Показать рассчитанные данные» в окне «Структурные данные и параметры пиков».
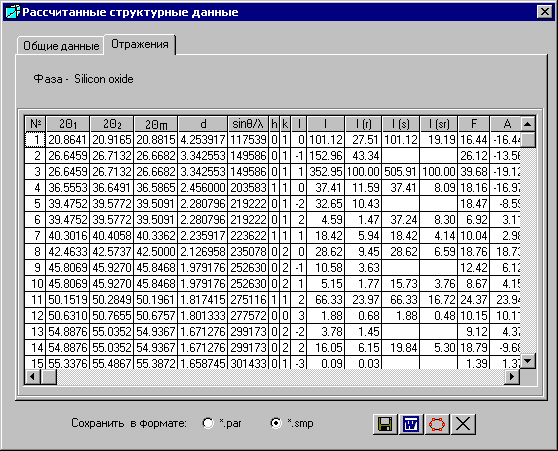
Если навести мышью на заголовок колонки, появится подсказка ее названия:
№ | - номер рефлекса. |
2q1, 2q2, 2qm | - углы отражений 2q для Ka1, Ka2, Kaср линий. |
d | - межплоскостные расстояния. |
sinq/l | - где q - угол отражения, l - длина волны излучения. |
hkl | - индексы отражений, которые можно отсортировать по возрастанию, если щелкнуть мышью на заголовке. Если индексы h будут иметь одинаковые значения, то сортируются индексы k. Если индексы h и k будут иметь одинаковые значения, то сортируются индексы l. |
I | - абсолютные интенсивности. |
I (r) | - относительные интенсивности. |
I (s) | - абсолютные суммарные интенсивности. Абсолютные интенсивности линий, расстояния между которыми меньше заданного разрешения, суммируются (см. окно «Условия эксперимента»). |
I (sr) | - относительные суммарные интенсивности. Относительные интенсивности линий, расстояния между которыми меньше заданного разрешения, суммируются (см. окно «Условия эксперимента»). |
F | - структурная амплитуда. |
A | - сумма косинусов (промежуточные значения в формуле для расчета структурной амплитуды). |
B | - сумма синусов (промежуточные значения в формуле для расчета структурной амплитуды). |
w | - полуширина Альфа1 пика. |
Ширину колонок можно менять курсором мыши. Некоторые колонки можно отсортировать по возрастанию, если щелкнуть мышью на заголовке.
Кнопки под таблицей описаны в главе «Общие данные».
5. Уточнение
5.1. Окно «Условия уточнения»
Окно «Условия уточнения» открывается при выборе кнопки ![]() - «Условия уточнения» в Главном окне.
- «Условия уточнения» в Главном окне.
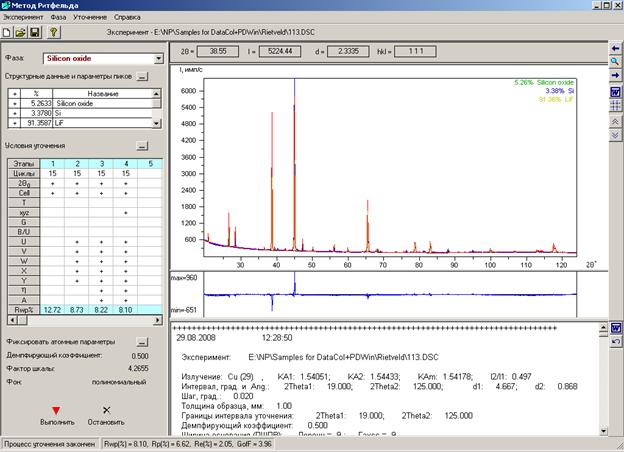
В окне «Условия уточнения» задают параметры для уточнения структурных данных.
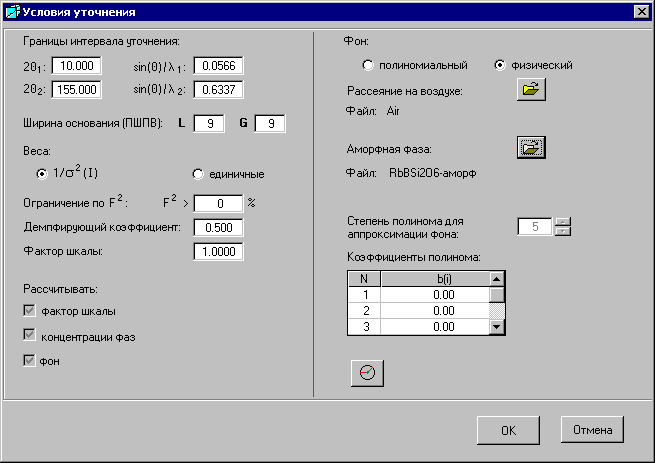
Границы интервала уточнения. Задают границы интервала уточнения. По умолчанию они соответствуют дифрактограмме снятого образца. Но их можно изменить. Щелкнув мышкой на любом другом вводимом на этой странице параметре, изменится и соответствующее углу значение sin(q) / l.
Ширина основания пика (ПШПВ) задается отдельно для функции Лоренца и для функции Гаусса. Угол, равный разности между положением максимума пика и половиной от указанного количества ПШПВ (полной ширины пика на половине его высоты), определяет начало пика. Угол, отстоящий от положения максимума на величину, равную половине от указанного ПШПВ, определяет конец пика. |
|
Веса. В процедуре уточнения используется два варианта весов. Один вариант – это единичные веса, когда все веса равны единице. Другой вариант – это величины, равные 1/s2(I), где I - экспериментальные интенсивности, s2 – дисперсии интенсивностей, которые рассчитываются исходя из того, что распределение интенсивностей подчиняется закону Пуассона. Активизированы по умолчанию.
Ограничение по F2. В процедуре уточнения используются только отражения со структурными факторами больше заданной величины в процентах.
Демпфирующий коэффициент – это коэффициент, на который умножаются приращения, полученные в процессе уточнения.
Фактор шкалы – для приведения экспериментальных и рассчитанных данных к одной шкале. По умолчанию фактор шкалы – 1.
Рассчитывать:
- фактор шкалы (можно задать вручную и не рассчитывать),
- концентрации фаз (введенные концентрации в Главном окне не будут меняться, если отменить галочку,
- фон (можно не рассчитывать).
![]() ВНИМАНИЕ! Для того чтобы отменить расчет фактора шкалы, концентраций фаз и расчет фона надо щелкнуть мышкой на галочке. Если галочка расчета не отменена, то независимо от того, какие значения фактора шкалы и концентраций были введены, эти значения будут уточняться после каждой итерации.
ВНИМАНИЕ! Для того чтобы отменить расчет фактора шкалы, концентраций фаз и расчет фона надо щелкнуть мышкой на галочке. Если галочка расчета не отменена, то независимо от того, какие значения фактора шкалы и концентраций были введены, эти значения будут уточняться после каждой итерации.
5.2. Ввод параметров для расчета фона
Окно «Условия уточнения» открывается при выборе кнопки ![]() - «Условия уточнения» в Главном окне.
- «Условия уточнения» в Главном окне.
В окне «Условия уточнения» задают параметры для расчета фона.
Фон – можно рассчитать либо:
- полиномиальный фон, используя степень аппроксимации фона,
- физический фон, используя файл рассеяния на воздухе и файл с аморфной фазой, если это необходимо.
![]() ВНИМАНИЕ! Файлы образца, рассеяния на воздухе и аморфная фаза должны быть сняты при одинаковых условиях эксперимента. Аморфная фаза должна быть того же химического состава, что и кристаллическая. Кристаллический образец может состоять из нескольких фаз, когда нет аморфной фазы.
ВНИМАНИЕ! Файлы образца, рассеяния на воздухе и аморфная фаза должны быть сняты при одинаковых условиях эксперимента. Аморфная фаза должна быть того же химического состава, что и кристаллическая. Кристаллический образец может состоять из нескольких фаз, когда нет аморфной фазы.
Рассеяние на воздухе ![]() - доступна только при выборе физического фона и открывает файл рассеяния на воздухе (съемка без образца и без кюветы).
- доступна только при выборе физического фона и открывает файл рассеяния на воздухе (съемка без образца и без кюветы).
Аморфная фаза ![]() - доступна только при выборе физического фона и открывает файл съемки аморфной фазы.
- доступна только при выборе физического фона и открывает файл съемки аморфной фазы.
Степень полинома для аппроксимации фона доступна только при выборе полиномиального фона. По умолчанию степень полинома равна 5. Можно выбрать от 1 до 100.
коэффициенты полинома появляются после проведения нулевого цикла. Количество их будет равно степени полинома. При выборе физического фона коэффициенты полинома будут равны нулю.
На примере видно, как прошел фон в образце с учетом рассеяния на воздухе и с учетом аморфной фазы:
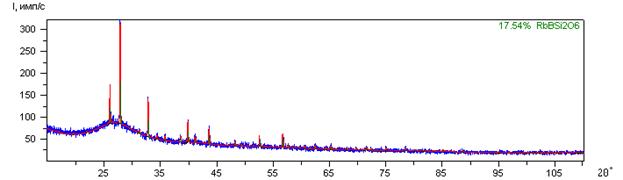
Рассчитывается процент кристаллической фазы в образце.
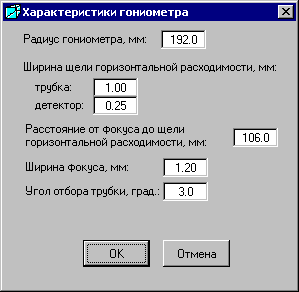
![]() - «Характеристики гониометра» открывает одноименное окно ««Характеристики гониометра», в котором по умолчанию заданы некие параметры, используемые для расчета компонент физического фона. Следует следить, чтобы щели горизонтальной расходимости соответствовали условиям съемки исследуемого образца, рассеяния на воздухе и аморфной фазы. По умолчанию заданы параметры гониометра для дифрактометра ДРОН-7.
- «Характеристики гониометра» открывает одноименное окно ««Характеристики гониометра», в котором по умолчанию заданы некие параметры, используемые для расчета компонент физического фона. Следует следить, чтобы щели горизонтальной расходимости соответствовали условиям съемки исследуемого образца, рассеяния на воздухе и аморфной фазы. По умолчанию заданы параметры гониометра для дифрактометра ДРОН-7.
5.3. Таблица с параметрами уточнения
В Главном окне представлена таблица с параметрами для уточнения. Параметры записаны в левом столбике таблицы (серый фон). Если навести мышкой на любой параметр, то высветится его полное название. 2q0 – нулевой угол детектора, Cell – параметры ячейки, T – текстурные параметры, xyz – атомные параметры, G – заселенность позиций, B/U – тепловые параметры, U, V, W, X, Y – параметры расчета полуширины пиков, h - доля функции Лоренца, A – общий параметр асимметрии. Для каждого этапа следует задать количество циклов и отметить мышью параметры, которые будут уточняться. Уточняемый параметр будет отмечаться крестиком «+». Для того чтобы перенести отмеченные крестиком «+» параметры в следующую колонку таблицы, следует щелкнуть на выбранном столбце в верхней строке «Этапы», а, затем, щелкнуть в верхней строке пустого столбца. |
|
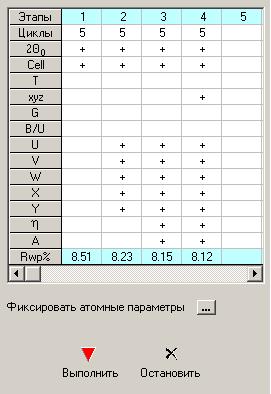
![]() ВНИМАНИЕ! Для каждой фазы создается своя таблица с уточняемыми параметрами. Только единственный параметр – это нулевой угол детектора «2q0» - будет общим для всех фаз.
ВНИМАНИЕ! Для каждой фазы создается своя таблица с уточняемыми параметрами. Только единственный параметр – это нулевой угол детектора «2q0» - будет общим для всех фаз.
Координаты атомов x, y, z, заселенности позиций G1, G2, G3 и тепловые параметры B/U можно зафиксировать, выбрав кнопку ![]() - «Фиксировать атомные параметры».
- «Фиксировать атомные параметры».
Условия уточнения можно сохранить, выбрав команду Сохранить как в меню «Уточнение». В файле с расширением *.ovr сохраняются условия уточнения, которые в данный момент выбраны в таблице уточнения. Условия уточнения, записанные в данном файле, можно применить для одной фазы, которая в данный момент отображается на экране, или для всех открытых фаз.
Для того чтобы начать процесс уточнения, следует выбрать кнопку ![]() - «Выполнить».
- «Выполнить».
Во время уточнения дифрактограммы образца и модели появляются вместе на одной картинке в рассчитанном процентном соотношении (см. Главное окно).
Процесс уточнения можно прервать, выбрав кнопку ![]() - «Остановить».
- «Остановить».
5.4. Окно «Фиксировать атомные параметры»
Окно «Фиксировать атомные параметры» открывается при выборе кнопки ![]() - «Фиксировать атомные параметры» в Главном окне.
- «Фиксировать атомные параметры» в Главном окне.
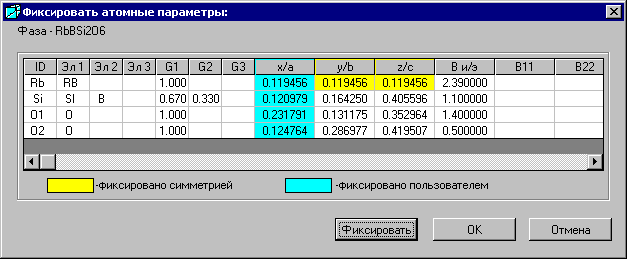
Параметры фиксируются следующим образом. Сначала их отмечают мышью, затем нажимают кнопку «Фиксация». Параметры закрасятся голубым цветом. Для того чтобы отменить фиксирование, действуют таким же образом.
«OK» - выходит в Главное окно.
«Отмена» - закрывает окно без запоминания внесенных данных.
5.5. Процесс уточнения
Процесс уточнения начинается после выбора кнопки ![]() - «Выполнить» в Главном окнеУто_нение_таблица_.
- «Выполнить» в Главном окнеУто_нение_таблица_.
Для уточнения параметров применяется метод оценки нелинейных параметров D. W.Marquardt (J. Soc. Indust. Appl. Math., 1963, vol. 11, № 2, June, p. 431-441).
На каждом этапе после каждого цикла уточняется фактор шкалы, концентрации фаз и расчет фона, но их уточнение можно отменить, тогда процесс уточнения пройдет немного быстрее.
Процесс уточнения и его результаты отражаются в статусной строке внизу экрана.
В процессе уточнения рассчитываются четыре R – фактора:
1. Взвешенный R-фактор: Rwp (%) = [Swi (Ii0 - Iic)2 / Swi I2i0]1/2
2. Профильный R-фактор: Rp (%) =SôIi0 - Iicô/ (SIi0)
3. Ожидаемый R-фактор: Re (%) = [ (N – P) / (Swi I2i0) ]1/2
4. Критерий G: GofF (%) = Swi (Ii0 - Iic)2 / (N – P) = (Rwp / Re )2
где wi = (s2i)-1 – веса, Iio – наблюдаемые интенсивности в каждой точке дифрактограммы, Iic - теоретические значения интенсивностей в каждой точке дифрактограммы, N – число точек дифрактограммы, P – число уточняемых параметров.
Полученный результат тем лучше, чем меньше значения R-факторов.
После выполнения каждого этапа в последней строке таблицы параметров уточнения на голубом фоне высвечиваются взвешенные R-факторы Rwp%.
![]() ВНИМАНИЕ! Если программа выдает сообщение о невозможности провести уточнение и требует изменить начальные параметры, то следует:
ВНИМАНИЕ! Если программа выдает сообщение о невозможности провести уточнение и требует изменить начальные параметры, то следует:
1. Проверить корректность начальных данных.
2. Уменьшить демпфирующий коэффициент, например, поставить 0.5.
3. Посмотреть, не пытаетесь ли Вы уточнять:
- все параметры элементарной ячейки (a, b, c) фазы, у которой сняты только пики с индексами отражения h00 и hk0,
- два параметра элементарной ячейки (a, b или a, c), а сняты только пики с индексами h00 для данной фазы.
4. Посмотреть, не стало ли значение h - доли составляющей Лоренца в функции Псевдо-Фойгта равным 1 для какой-либо фазы. В этом случае не следует уточнять параметры полуширины пиков U, V, W для этой фазы.
6. Результаты
Полученные результаты уточнения высвечиваются в Главном окне в правой нижней панели.
Отображаются начальные данные и уточненные данные на каждом этапе и цикле уточнения.

![]() - «Копировать текст» открывает текстовый редактор MSWord с результатами расчета.
- «Копировать текст» открывает текстовый редактор MSWord с результатами расчета.
![]() ВНИМАНИЕ! Если MSWord на компьютере не установлен, то следует открыть редактор WordPad и выбрать кнопку
ВНИМАНИЕ! Если MSWord на компьютере не установлен, то следует открыть редактор WordPad и выбрать кнопку ![]() - «Вставить».
- «Вставить».
![]() - «Очистить текстовое окно» уничтожает все рассчитанные данные.
- «Очистить текстовое окно» уничтожает все рассчитанные данные.
Геометрия
1. Геометрические связи
Расчет геометрических параметров для выбранной фазы можно сделать в окне «Геометрия для ***», выбрав инструмент ![]() - «Геометрия» в окне «Рассчитанные структурные данные/Общие данные».
- «Геометрия» в окне «Рассчитанные структурные данные/Общие данные».

На верхних панелях данного окна представлены атомы, входящие в структуру открытой фазы, и их координаты.
Два атома называются связанными, если расстояния между ними больше некоторого минимального расстояния и меньше расстояния, определяемого как сумма радиусов сфер, проведенных вокруг каждого из атомов и некоторой положительной дельты. Минимальное расстояние Dmin и дельта D задаются пользователем. По умолчанию их значения - Dmin=0.5 и D=0.2. Соответствующие радиусы для каждого атома выбираются из файла user. par. В дальнейшем все расстояния и углы считаются между атомами, удовлетворяющими этому условию.
В расчете расстояний от атомов до плоскости участвуют веса атомов, которые могут быть единичными, т. е. равными для всех атомов, или соответствовать количеству электронов в атоме.
В приведенном примере выбрана фаза кальциевый хлорванадат Ca2VO4Cl, содержащая два сорта базисных атома кальция – Ca1 и Ca2, по одному атому хлора Cl и ванадия V и два атома кислорода – O1 и O2, которые идентифицируются в колонке ID.
Колонка «Флаг» на левой панели позволяет выбрать сразу все атомы данного сорта. Например, если бы мы выбрали атом Ca в колонке «Флаг» на левой панели, в колонке «Флаг» на правой панели автоматически выбрались бы все атомы Ca (Ca1 и Ca2).
Радиусы сфер вокруг атомов Ca, Cl, V, O соответственно равны 1.97; 1.34; 0.7; 1.35 и записаны в колонке R.
![]() - «Рассчитать» осуществляет расчет геометрических параметров, отмеченных галочкой.
- «Рассчитать» осуществляет расчет геометрических параметров, отмеченных галочкой.
![]() - «Копировать» запоминает и копирует полученные данные в текстовый редактор MSWord.
- «Копировать» запоминает и копирует полученные данные в текстовый редактор MSWord.
![]() ВНИМАНИЕ! Если MSWord на компьютере не установлен, то следует открыть редактор WordPad и выбрать кнопку
ВНИМАНИЕ! Если MSWord на компьютере не установлен, то следует открыть редактор WordPad и выбрать кнопку ![]() - «Вставить».
- «Вставить».
![]() - «Закрыть» закрывает данное окно без запоминания результатов.
- «Закрыть» закрывает данное окно без запоминания результатов.
2. Расчет межатомных расстояний и валентных углов
Расчет межатомных расстояний и валентных углов можно сделать в окне «Геометрия для ***», выбрав инструмент ![]() - «Геометрия» в окне «Рассчитанные структурные данные/Общие данные».
- «Геометрия» в окне «Рассчитанные структурные данные/Общие данные».
Для расчета межатомных расстояний и валентных углов следует активизировать флажок «Расстояния и валентные углы» и выбрать атомы в колонке «Флаг», между которыми будут рассчитываться расстояния и углы.
В предложенном примере выбраны атомы Са2 и O2.
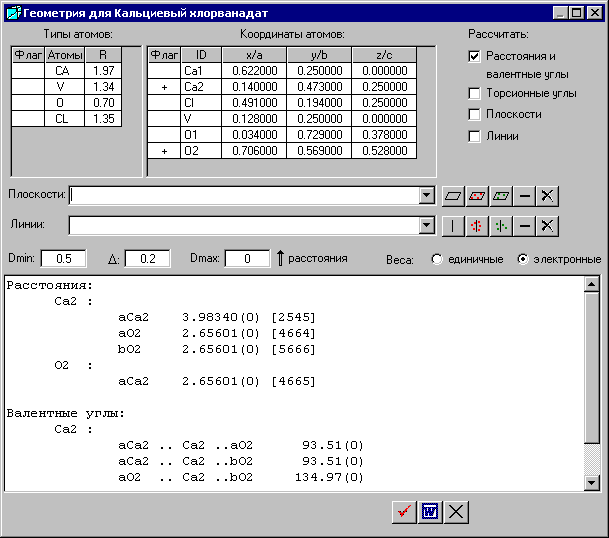
После выбора ![]() - «Рассчитать» на белом фоне нижней панели появляются следующие данные:
- «Рассчитать» на белом фоне нижней панели появляются следующие данные:
1. Расстояния между выбранными атомами
Программа рассчитывает расстояния, и в круглых скобках указаны погрешности межатомных расстояний, которые рассчитываются из погрешностей координат атомов. В данном случае погрешности координат атомов были равны нулю, поэтому погрешности межатомных расстояний также получились равными нулю.
Если атому предшествует буква, это значит, что на базисные атомы мы подействовали преобразованиями симметрии. Например, если атому предшествует буква «a», то на базисные атомы мы подействовали 1-ым оператором симметрии, если «b», то 2-ым оператором симметрии и т. д. Номера операторов соответствуют номеру эквивалентной позиции.
В квадратных скобках даны числовые символы, состоящие из номера эквивалентной позиции (оператора симметрии) и величины трансляционного сдвига. С их помощью определяют координаты соответствующего атома. Например, межатомное расстояние между атомом Ca2 и другим атомом aCa2 равно 3.98340 с погрешностью, равной нулю. Координаты атома aCa2 определяются из базисной позиции симметричным преобразованием под номером 2, которые можно посмотреть в окне «Результаты» на вкладке «Структурные данные». Затем, к эквивалентной позиции следует прибавить трансляционные компоненты 0 –1 0 вдоль направлений x, y и z соответственно. Число 5 означает 0-ю трансляцию, 4 следует рассматривать как x - 1, а 6 – как x + 1 и т. д.
2. Валентные углы между выбранными атомами
Рассмотрим угол, образованный атомами aCa2, Ca2, aO2 с атомом Ca2 в вершине.
В круглых скобках указаны погрешности валентных углов, которые рассчитываются из погрешностей координат атомов. В данном случае погрешности координат атомов были равны нулю, поэтому погрешности валентных углов также получились равными нулю.
2. Расчет торсионных углов
Расчет торсионных углов можно сделать в окне «Геометрия для ***», выбрав инструмент ![]() - «Геометрия» в окне «Рассчитанные структурные данные/Общие данные».
- «Геометрия» в окне «Рассчитанные структурные данные/Общие данные».
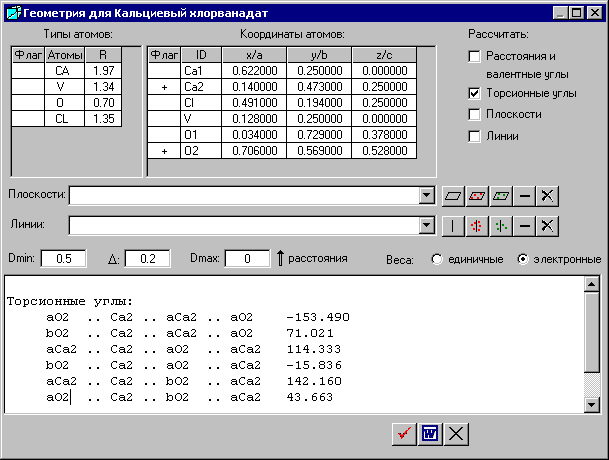
Для расчета торсионных углов следует активизировать флажок «Торсионные углы» и выбрать атомы в колонке «Флаг», между которыми будут рассчитываться торсионные углы. В предложенном примере выбраны атомы Са2 и O2.

|
Из за большого объема этот материал размещен на нескольких страницах:
1 2 3 |
