

Партнерка на США и Канаду по недвижимости, выплаты в крипто
- 30% recurring commission
- Выплаты в USDT
- Вывод каждую неделю
- Комиссия до 5 лет за каждого referral
НАУЧНО-ПРОИЗВОДСТВЕННОЕ ПРЕДПРИЯТИЕ
«БУРЕВЕСТНИК», ОАО
Программа
Метод Ритфельда – Rietveld
Руководство оператора
Санкт-Петербург
2008 г.
СОДЕРЖАНИЕ
Введение.. 3
Строка меню.... 4
1. Меню «Эксперимент». 4
2. Меню «Фаза». 4
3. Меню «Уточнение». 4
4. Меню «Справка.. 4
Панель инструментов.. 5
Работа по программе.. 6
1. Выбор файла эксперимента.. 6
1.1. Окно «Открыть файл эксперимента». 6
1.2. Окно «Условия эксперимента». 7
1.3. Дифрактограмма эксперимента. 8
2. Выбор файла фазы... 9
2.1. Окно «Открыть файл фазы». 9
3. Окно «Структурные данные и параметры пиков». 10
3.1. Ввод общих и атомных данных для расчета структурных данных. 10
3.2. Пространственная группа. 11
3.3. Параметры элементарной ячейки. 11
3.4. Окно «Химический состав». 12
3.5. Окно «Атомные данные для фазы». 13
3.6. Окно «Преобразование координат». 15
3.7. Данные атомного рассеяния. 16
3.8. Ввод параметров пиков для расчета полуширины линий. 17
4. Окно «Рассчитанные структурные данные». 19
4.1. Общие данные. 19
4.2. Окно «Сохранить как» - сохранение в форматах *.par и *.smp. 20
4.3. Отражения. 21
5. Уточнение.. 23
5.1. Окно «Условия уточнения». 23
5.2. Ввод параметров для расчета фона. 25
5.3. Таблица с параметрами уточнения. 26
5.4. Окно «Фиксировать атомные параметры». 27
5.5. Процесс уточнения. 27
6. Результаты... 28
Геометрия.. 29
1. Геометрические связи.. 29
2. Расчет межатомных расстояний и валентных углов.. 30
2. Расчет торсионных углов.. 31
3. Расчет плоскости, проведенной через группу атомов.. 32
4. Расчет линии, проведенной через группу атомов.. 35
Структура файлов.. 38
1. Файл эксперимента для ДРОН-7. 38
2. Файл эксперимента для ДРОН-4. 39
3. Текстовый файл эксперимента.. 39
Введение
Программа уточнения атомной структуры методом Ритфельда предназначена для уточнения структуры/структур однофазного или полифазного образца по экспериментальным порошковым дифрактометрическим данным.
Исходными данными являются дифрактограмма образца и структурные данные фаз, входящих в исследуемый образец. Экспериментальные дифрактометрические данные могут быть введены из файлов, полученных на дифрактометрах ДРОН-7, ДРОН-6, ДРОН-4, STOE, HUBER или из текстовых файлов в формате DBW, содержащих информацию об интенсивностях и углах 2q (d), полученную на других поликристальных дифрактометрах. В качестве входных файлов со структурными данными фаз программа может использовать файлы форматов CSD, SHELX, СIF, ICSD.
Уточнение включает в себя уточнение фона (фон аппроксимируется степенным полиномом, степень которого выбирается пользователем), уточнение фактора шкалы и расчет концентрационных соотношений. Стратегия уточнения определяется таблицей, в которой пользователь указывает параметры, уточняемые на различных этапах процедуры. В процессе уточнения, на любом его этапе, исходя из промежуточных результатов, можно изменять стратегию уточнения. Для уточнения используется весовой нелинейный метод наименьших квадратов (метод Маркварта).
Для уточнения доступны следующие параметры: нулевой угол детектора, параметры элементарной ячейки, атомные и тепловые параметры, профильные параметры, концентрационные соотношения и коэффициенты профильного полинома. Под атомными параметрами здесь понимаются координаты и тепловые (изотропные/анизотропные) параметры атомов, коэффициенты заселенности позиций и текстурные параметры.
Из особенностей программы следует отметить возможность ввода и дальнейшего уточнения различных значений коэффициентов U, V, W, X и Y, которые определяют полуширины пиков через функции Гаусса и Лоренца не только для разных фаз, но и для рефлексов с разными типами индексов в одной фазе. Кроме того, пользователь может, в качестве начального приближения, выбрать соотношение Гаусс/Лоренц в функции псевдо-Фойгта и параметр асимметрии функции.
Очень часто в процессе уточнения профильных параметров возникает вопрос о том, как выбрать границы пика. Практически 5-6 полуширин оказывается вполне приемлемой величиной. Однако хорошо известно, что функция Гаусса спадает до фоновых значений гораздо быстрее, чем функция Лоренца. Поэтому, для сокращения времени вычислений, в программе предусмотрена возможность раздельного выбора ширины оснований пиков для составляющих Гаусса и Лоренца.
Для предотвращения разноса уточняемых параметров возможно использование демпфирующего фактора.
Фиксация и связи между позиционными и тепловыми параметрами атомов, находящихся в частных позициях, генерируются автоматически в процессе их ввода. Пользовательская фиксация атомных параметров и наложение связей между ними осуществляются на этапе ввода информации об индивидуальной фазе.
На этапе выбора стратегии возможно разрешить/запретить уточнение всех позиционных и/или всех тепловых параметров и/или всех параметров заселенности, значение которых разрешено уточнять на этапе ввода информации об индивидуальной фазе.
Профильные параметры U, V, W, X, Y, Гаусс/Лоренц соотношение и параметр асимметрии можно разрешить/запретить индивидуально для каждой фазы.
Программа позволяет рассчитать межатомные расстояния и валентные углы, торсионные углы и уравнения плоских и линейных фрагментов структуры и углов между ними, а также уточнить координаты атомов с помощью рядов Фурье.
Программу можно использовать для проведения количественного фазового анализа по известной атомной структуре фаз.
В программе используются понятия «Эксперимент» и «Фаза». Под словом «Эксперимент» подразумевается дифрактограмма одной фазы или смеси из нескольких фаз. Под словом «Фаза» подразумеваются атомные структурные данные для фаз, входящих в образец.
- в системе Windows 2000 папку «Язык и стандарты», на панели «Числа» поменять разделитель целой и дробной частей числа на точку,
- в системе Windows XP папку «Язык и региональные стандарты», кнопку «Настройка», в окне «Настройка региональных параметров» поменять разделитель целой и дробной части на точку,
- в системе Vista папку «Язык и региональные стандарты», кнопку «Изменить этот формат», в окне «Настройка региональных параметров» поменять разделитель целой и дробной части на точку.
Строка меню
1. Меню «Эксперимент»
Открывает команды:
Открыть - открывает окно «Открыть файл эксперимента» (см. гл. «Окно «Открыть файла эксперимента»»). Команда становится не доступна после выбора файла эксперимента. Условия - доступна после открытия файла эксперимента и открывает окно «Условия эксперимента» (см гл. «Окно «Условия эксперимента»»). |
|
Выход - закрывает программу.
2. Меню «Фаза»
Открывает команды:
Новая фаза - доступна после выбора файла эксперимента и открывает окно «Структурные данные и параметры пиков» (см. гл. «Пространственная группа»). Открыть - доступна после выбора файла эксперимента и открывает окно «Открыть файл фазы» (см. гл. «Окно «Открыть файл фазы»»). |
|

Закрыть - закрывает любую фазу, выбранную из списка ранее открытых фаз.
3. Меню «Уточнение»
Открывает команды:
Открыть - открывает файл с расширением *.ovr. Это ранее запомненный файл с условиями уточнения, который можно прочитать любым текстовым редактором, например, «Блокнотом». |
|
Условия уточнения, записанные в данном файле, можно применить для одной фазы, которая в данный момент отображается на экране, или для всех открытых фаз.
Сохранить как - открывает окно «Сохранить как», где задают имя файла с расширением *.ovr. В файле запоминаются условия уточнения, которые в данный момент выбраны в таблице уточнения.
4. Меню «Справка
Открывает команды:
Вызов справки - открывает справочную систему с информацией о работе программы. О программе - открывает информационное окно о версии программы и данные о разработчиках. |
|

Панель инструментов
![]() - «Открыть новую фазу» доступен после выбора файла эксперимента и открывает окно «Структурные данные и параметры пиков» (см. гл. «Пространственная группа»). Данный инструмент дублирует команду Новая фаза в меню «Фаза».
- «Открыть новую фазу» доступен после выбора файла эксперимента и открывает окно «Структурные данные и параметры пиков» (см. гл. «Пространственная группа»). Данный инструмент дублирует команду Новая фаза в меню «Фаза».
![]() - «Открыть файл фазы» доступен после выбора файла эксперимента и открывает окно «Открыть файл фазы» (см. гл. «Окно «Открыть файл фазы»»). Данный инструмент дублирует команду Открыть в меню «Фаза».
- «Открыть файл фазы» доступен после выбора файла эксперимента и открывает окно «Открыть файл фазы» (см. гл. «Окно «Открыть файл фазы»»). Данный инструмент дублирует команду Открыть в меню «Фаза».
![]() - «Сохранить данные для фазы» доступен после выбора файла фазы с рассчитанными структурными данными или после выбора кнопок
- «Сохранить данные для фазы» доступен после выбора файла фазы с рассчитанными структурными данными или после выбора кнопок ![]() - «Рассчитать и сохранить данные» и
- «Рассчитать и сохранить данные» и ![]() - «Закрыть» в окне «Структурные данные и параметры пиков». Сохраняет файл фазы под тем же именем в том же каталоге. Образуются три файла: *.par – файл со структурными данными, *.hkp – файл с рассчитанными интенсивностями и структурными факторами и *.spr – файл с характеристиками пиков.
- «Закрыть» в окне «Структурные данные и параметры пиков». Сохраняет файл фазы под тем же именем в том же каталоге. Образуются три файла: *.par – файл со структурными данными, *.hkp – файл с рассчитанными интенсивностями и структурными факторами и *.spr – файл с характеристиками пиков.
![]() - «Справка» открывает справочную систему с информацией о работе программы. Данный инструмент дублирует команду Вызов справки в меню «Справка».
- «Справка» открывает справочную систему с информацией о работе программы. Данный инструмент дублирует команду Вызов справки в меню «Справка».
Работа по программе
1. Выбор файла эксперимента
1.1. Окно «Открыть файл эксперимента»
Команда Открыть в меню «Эксперимент» открывает окно «Открыть файл эксперимента»:
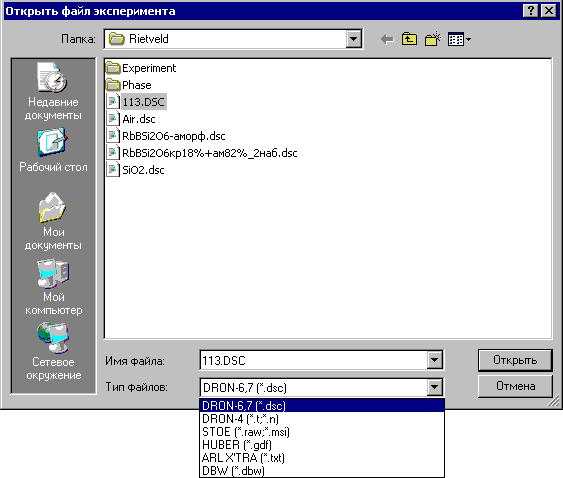
В данном окне по умолчанию открываются файлы с расширением *.dsc, полученные при съемке на дифрактометрах ДРОН-6 или ДРОН-7, но можно выбрать файлы эксперимента, полученные на других дифрактометрах:
DRON-4 - файлы с расширением dscr. t или dscr. n, полученные при съемке на дифрактометре ДРОН-4,
STOE – файлы с расширением *.raw и *.msi, полученные при съемке на аппарата фирмы STOE,
HUBER - файлы с расширением *.gdf, полученные при съемке на аппарата фирмы HUBER,
ARLX’TRA - файлы с расширением *.txt, текстовые файлы эксперимента,
DBW - файлы с расширением *.dbw в широко используемом разными фирмами формате.
После выбора файла эксперимента сразу открывается окно «Условия эксперимента».
1.2. Окно «Условия эксперимента»
Команда Условия в меню «Эксперимент» доступна после открытия файла эксперимента и открывает окно «Условия эксперимента».
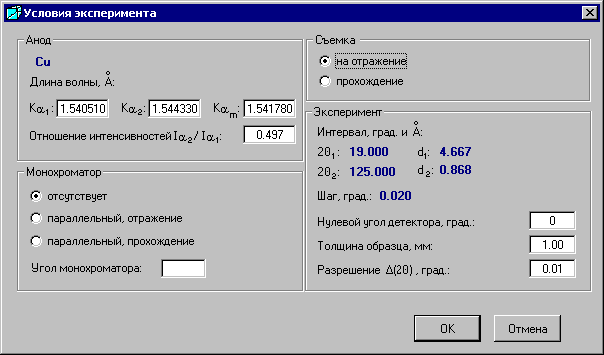
Анод, интервал и шаг съемки взяты из файла съемки и не подлежат изменению. Когда снято несколько интервалов, то будут отображаться начало первого интервала 2q1 и конец последнего 2q2, а рядом - соответствующие углам межплоскостные расстояния d.
Можно изменить значение длины волны, отношение интенсивностей, условия съемки с монохроматором, нулевой угол детектора, толщину образца и разрешение.
Разрешение используется лишь для графического представления расчетной (теоретической) дифрактограммы. Два рефлекса будут изображаться одним пиком, если угловое расстояние между ними меньше заданного разрешения, т. е. пики с расстоянием меньшим, чем угол разрешения, суммируются.
При выборе кнопки «OK» внесенные изменения запоминаются. При выборе кнопки «Отмена» данные остаются неизменными.
После закрытия данного окна на странице исходных данных открывается дифракционная картина выбранного файла эксперимента.
Название открытого образца записывается в строку панели инструментов.
1.3. Дифрактограмма эксперимента
Дифрактограмма эксперимента появляется после выбора файла эксперимента (команда Открыть в меню «Эксперимент»).
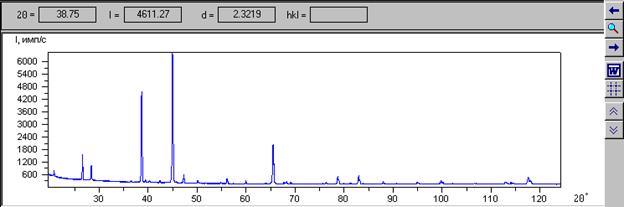
Курсор мыши указывает угол 2Q и интенсивность I. Угол 2Q автоматически пересчитывается в d – межплоскостное расстояние. Для расчета используется формула Брегга-Вульфа: nl=2d(hkl)sinQ. В данной программе порядок отражения n равен единице и используется длина волны l для линии Ka1.
Дифракционную картину можно посмотреть подробнее, растянув мышью нужный кусок интервала. Количество точек, попадающих в растягиваемую часть интервала, не должно быть меньше двух.
В работе с дифракционной картиной помогают следующие инструменты:
![]() - «Масштаб 1:1» возвращает дифракционную картину в первоначальный масштаб. Инструмент становится доступен после растяжения интересующего участка дифракционной картины на весь экран.
- «Масштаб 1:1» возвращает дифракционную картину в первоначальный масштаб. Инструмент становится доступен после растяжения интересующего участка дифракционной картины на весь экран.
![]()
![]() - «Двигать влево (вправо)» двигает дифракционную картину без изменения масштаба.
- «Двигать влево (вправо)» двигает дифракционную картину без изменения масштаба.
![]() - «Копировать» открывает текстовый редактор MSWord с дифракционной картиной, которая в текущий момент отображается на экране. Если ранее был открыт документ MSWord, то дифракционная картина будет внесена в тот же документ.
- «Копировать» открывает текстовый редактор MSWord с дифракционной картиной, которая в текущий момент отображается на экране. Если ранее был открыт документ MSWord, то дифракционная картина будет внесена в тот же документ.
![]() ВНИМАНИЕ! Если MSWord на компьютере не установлен, то следует открыть редактор WordPad и выбрать кнопку
ВНИМАНИЕ! Если MSWord на компьютере не установлен, то следует открыть редактор WordPad и выбрать кнопку ![]() - «Вставить».
- «Вставить».
![]() - «Масштабная сетка» убирает/показывает масштабную сетку.
- «Масштабная сетка» убирает/показывает масштабную сетку.
![]() - «Увеличить высоту рисунка» растягивает дифрактограмму в вертикальном направлении по шагам.
- «Увеличить высоту рисунка» растягивает дифрактограмму в вертикальном направлении по шагам.
![]() - «Уменьшить высоту рисунка» сокращает дифрактограмму в вертикальном направлении по шагам.
- «Уменьшить высоту рисунка» сокращает дифрактограмму в вертикальном направлении по шагам.
2. Выбор файла фазы
2.1. Окно «Открыть файл фазы»
Команда Открыть в меню «Фаза» доступна после выбора файла эксперимента и открывает окно «Открыть файл фазы»:
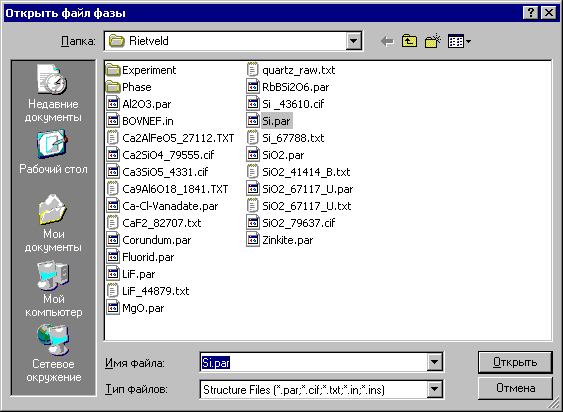
В данном окне можно выбрать следующие файлы фазы со структурными данными:
CSD - файлы с расширением *.par для комплексов PDWin и CSD,
SHELX - файлы с расширением *.in и *.ins,
ICSD (Inorganic Chemistry Structure Database) - файлы с расширением *.txt,
CIF (Crystallographic Information File) - файлы с расширением *.cif.
3. Окно «Структурные данные и параметры пиков»
3.1. Ввод общих и атомных данных для расчета структурных данных
Окно «Структурные данные и параметры пиков» открывается после выбора команд Новая фаза или Открыть в меню «Фаза», которые становятся доступны после выбора файла эксперимента.
В первом случае (команда Новая фаза) данные вводятся, во втором случае (команда Открыть) данные читаются из файлов.
Если открыт файл фазы, то окно «Структурные данные и параметры пиков» будет заполнено. Если выбрана новая фаза, то следует задать ее название в строке «Фаза» и внести начальные данные для расчета ее структурных и дифрактометрических данных.
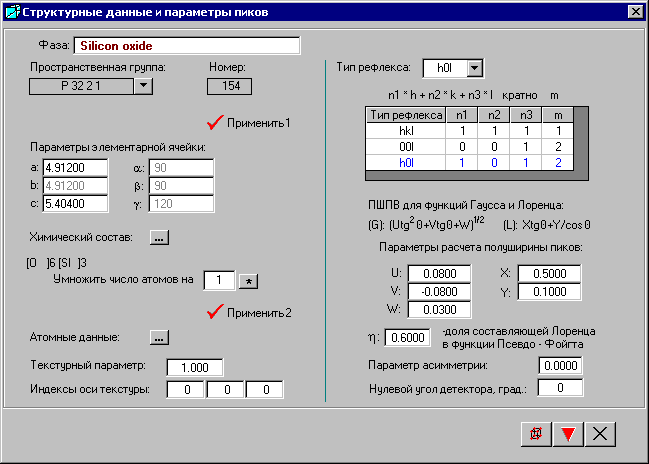
Из раскрывающегося списка выбирают пространственную группу в любой возможной установке (см. главу «Пространственная группа»).
После выбора кнопки ![]() - «Применить 1» генерируются матрицы симметрии, и появляется возможность ввода параметров элементарной ячейки (см. главу «Параметры элементарной ячейки»).
- «Применить 1» генерируются матрицы симметрии, и появляется возможность ввода параметров элементарной ячейки (см. главу «Параметры элементарной ячейки»).
Химический состав фазы вводят в окне «Химический состав» (см. главу «Окно «Химический состав»»), выбрав кнопку ![]() - «Химический состав».
- «Химический состав».
Чтобы быть уверенным, что введенная химическая формула верна, полезно сравнить рассчитанную плотность в окне «Рассчитанные структурные данные/Общие данные» с реальным значением плотности. Если они отличаются, необходимо уменьшить или увеличить количество атомов для каждого элемента в строке «Умножить число атомов на» и выбрать кнопку ![]() . Кроме целых чисел можно вводить и дробные числа, например: 0.5 или 2/3.
. Кроме целых чисел можно вводить и дробные числа, например: 0.5 или 2/3.
Далее следует выбрать кнопки ![]() - «Применить 2» и
- «Применить 2» и ![]() - «Атомные данные» для ввода атомных данных (см. главу «Окно «Атомные данные для фазы»»).
- «Атомные данные» для ввода атомных данных (см. главу «Окно «Атомные данные для фазы»»).
![]() ВНИМАНИЕ! После выбора кнопки
ВНИМАНИЕ! После выбора кнопки ![]() - «Применить 2» могут появиться дополнительные сообщения (см. главу «Данные атомного рассеяния»).
- «Применить 2» могут появиться дополнительные сообщения (см. главу «Данные атомного рассеяния»).
При наличии текстуры (например, преимущественной ориентации пластинчатой или игольчатой формы частиц вещества) значение текстурного параметра вводят вдоль какого-нибудь кристаллографического направления, определяющего текстуру. Для кристаллов пластинчатой формы текстурный параметр должен быть меньше единицы, для кристаллов игольчатой формы - больше единицы. В программе используется однопараметрическая модель текстуры.
3.2. Пространственная группа
Для заданной фазы из раскрывающегося списка выбирают пространственную группу в любой возможной установке. На основе символа пространственной группы по Герману-Могену определяется сингония и номер пространственной группы по Интернациональным таблицам. |
|
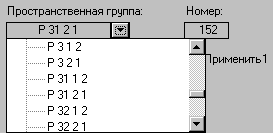
После выбора кнопки ![]() - «Применить 1» генерируются матрицы симметрии, и появляется возможность ввода параметров элементарной ячейки (см. главу «Параметры элементарной ячейки»).
- «Применить 1» генерируются матрицы симметрии, и появляется возможность ввода параметров элементарной ячейки (см. главу «Параметры элементарной ячейки»).
3.3. Параметры элементарной ячейки
После выбора пространственной группы и кнопки ![]() - «Применить 1» в окне «Структурные данные и параметры пиков» вводят параметры элементарной ячейки. Линейные параметры не должны быть менее 1.2 ангстрем. Ввести можно лишь независимые параметры ячейки. Остальные присваиваются автоматически.
- «Применить 1» в окне «Структурные данные и параметры пиков» вводят параметры элементарной ячейки. Линейные параметры не должны быть менее 1.2 ангстрем. Ввести можно лишь независимые параметры ячейки. Остальные присваиваются автоматически.
№ | Сингония | Вводимые параметры | Параметры, определенные из сингонии |
1 | Кубическая | a | a = b = c, a = b = g = 90 град. |
2 | Тетрагональная | a, c | a = b, a = b = g = 90 град. |
3 | Гексагональная и тригональная | a, c | a = b, a = b = 90 град., g = 120 град. |
4 | Тригональная (ромбоэдрическая) | a, a | a = b = c, a = b = g |
5 | Ромбическая | a, b, c | a = b = g = 90 град. |
6 | Моноклинная | a, b, c, b | a = g = 90 град. |
7 | Триклинная | a, b, c, a, b, g | - |
3.4. Окно «Химический состав»
Для того чтобы ввести химический состав фазы, следует выбрать кнопку ![]() - «Химический состав» в окне «Структурные данные и параметры пиков».
- «Химический состав» в окне «Структурные данные и параметры пиков».
Появится таблица химических элементов в окне «Химический состав», где выбирают элементы, щелкая на них мышью.
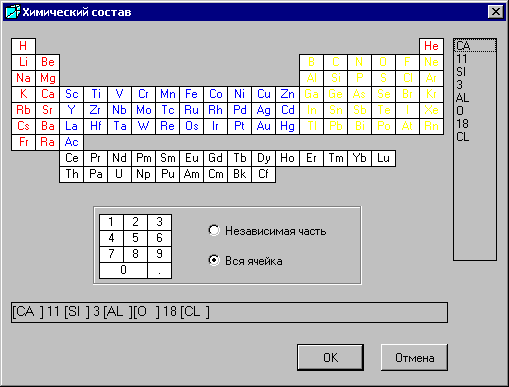
Элемент заносится в правую колонку для редактирования и в нижнюю строку для просмотра химической формулы. Формула может содержать не более 16 элементов. Число атомов вводится с помощью таблицы цифр.
· Независимая часть – для ввода числа атомов данного элемента на независимую часть элементарной ячейки. Цифры имеют отрицательные значения.
· Вся ячейка – для ввода числа атомов данного элемента для всей элементарной ячейки. Цифры имеют положительные значения.
Приведем пример ввода химического состава фазы алинит: Ca 11 [ ( Si 0.75 Al 0.25) O 4 ] 4 O 2 Cl.
Основные моменты ввода:
1. Количество атомов на всю ячейку вводится со знаком «+», и в этом случае количество атомов (в нашем примере это Cl и Al), равных 1 можно не вводить. Если выбрана радио-кнопка «Независимая часть», то количество атомов на независимую часть ячейки вводится со знаком «-».
2. Атомы кремния Si и алюминия Al занимают одинаковые позиции. Соотношение между ними 3:1, общее число атомов равно 4 (см. формулу), поэтому Si = 3 и Al = 1.
3. Атом кислорода O в формуле встречается два раза, но вводить его нужно только один раз, суммируя все значения числа атомов. Общее число атомов кислорода равно 18.
В правой колонке можно отредактировать химический состав фазы. Для этого следует выбрать мышью атом или его число, появится окно со следующими командами:
Удалить - удаляет выбранную строку,
Вставить - вставляет строку выше выбранной строки,
Очистить - удаляет всю колонку.
После выбора кнопки «OK» количество атомов в химической формуле на странице исходных данных может пересчитываться на всю ячейку, если была выбрана радио-кнопка «Независимая часть». Количество атомов на независимую часть ячейки умножается на кратность точки общего положения.
Чтобы быть уверенным, что введенная химическая формула верна, полезно сравнить рассчитанную плотность в окне «Рассчитанные структурные данные/Общие данные» с реальным значением плотности. Если они отличаются, необходимо уменьшить или увеличить количество атомов для каждого элемента в строке «Умножить число атомов на» в окне «Структурные данные и параметры пиков» и выбрать кнопку ![]() . Кроме целых чисел можно вводить и дробные числа, например: 0.5 или 2/3.
. Кроме целых чисел можно вводить и дробные числа, например: 0.5 или 2/3.
3.5. Окно «Атомные данные для фазы»
Атомные данные вводят в окне «Структурные данные и параметры пиков», выбрав кнопку ![]() - «Атомные данные». Появится окно «Атомные данные для фазы - *».
- «Атомные данные». Появится окно «Атомные данные для фазы - *».
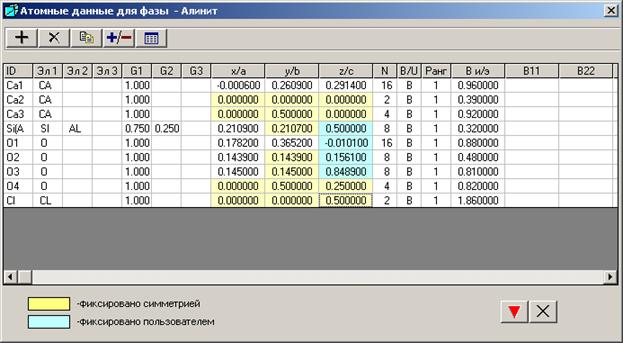
Параметры, зафиксированные элементами симметрии, закрасятся желтым цветом сразу после выбора кнопки «Применить». Пользователь может зафиксировать необходимые параметры в окне «Фиксировать атомные параметры», выбрав соответствующую кнопку ![]() в Главном окне.
в Главном окне.
Ширину всех колонок можно изменить курсором мыши.
Названия колонок:
ID - вводят идентификатор атома. Идентификатор может состоять из любого сочетания цифр и букв, но запомнятся только 4 символа. Если последний символ был цифрой, то при вводе новой строки атому автоматически присваивается следующий порядковый номер. Можно отсортировать атомы, если щелкнуть на заголовке колонки.
Эл 1, Эл 2, Эл 3 - вводят название атома из открывающего списка, который формируется в соответствии с введенным химическим составом. Если в фазе более чем один тип атома в одной позиции, то заполняют колонки «Эл 2» и «Эл 3». Всего может быть не более трех атомных типов в одной позиции.
G1, G2, G3 - вводят заселенности для имеющихся типов атомов, которых может быть три. Если атом занимает 100% позицию, заселенность для него будет равна 1. Программа позволяет работать с различными изоморфными смесями атомов в одних позициях. Заселенность позиции может быть любым значением меньше 1. Сумма заселенностей различных атомов в одной позиции может быть меньше, чем 1 (100%), так же как и больше 1 (100%). (Очевидно, что последнее не имеет большого физического смысла).
Пример: позиция занята тремя типами атомов: Ca, Mg и Fe. Известно, что соотношение между числом атомов Mg и Fe равно 3:1. И, наконец, общая заселенность позиции Ca составляет 60%. Поэтому,
G(Ca) = 0.6
G(Mg) + G(Fe) = 0.4 G(Mg) = 0.3
G(Mg) / G(Fe) = 3/1 G(Fe) = 0.1
x/a, y/b, z/c - вводят координаты атомов в долях оси. Значения их параметров имеют десятичные значения.
N - содержит кратности позиции для каждого атомного типа, которые рассчитываются программой после выбора кнопки ![]() - «Рассчитать и сохранить данные».
- «Рассчитать и сохранить данные».
B/U - если щелкнуть на заголовке, то поменяются единицы измерения тепловых параметров.
Ранг - определяет: 1 - изотропную температурную поправку, 2 – анизотропную температурную поправку.
B и/э - вводят изотропную или анизотропную (эквивалентную) температурную поправку:
B и/э = 1/3 (B11 + B22 + B33 + B12 + B13 + B23)
U и/э = 1/3 (U11 + U22 + U33 + U12 + U13 + U23)
По умолчанию ее величина равна единице.
B11, B22, B33, B12, B13, B23 - для ввода анизотропных температурных поправок.
Панель инструментов в окне «Атомные данные для фазы - *»:
![]() - «Добавить атом» вводит новую строку.
- «Добавить атом» вводит новую строку.
![]() - «Удалить атом» удаляет строку, если предварительно ее отметить мышью в первой колонке ID.
- «Удалить атом» удаляет строку, если предварительно ее отметить мышью в первой колонке ID.

|
Из за большого объема этот материал размещен на нескольких страницах:
1 2 3 |
