
Партнерка на США и Канаду по недвижимости, выплаты в крипто
- 30% recurring commission
- Выплаты в USDT
- Вывод каждую неделю
- Комиссия до 5 лет за каждого referral
транспортная — через мембрану происходит транспорт веществ в клетку и из клетки. Транспорт через мембраны обеспечивает: доставку питательных веществ, удаление конечных продуктов обмена, секрецию различных веществ, создание ионных градиентов, поддержание в клетке оптимального pH и концентрации ионов, которые нужны для работы клеточных ферментов.
Частицы, по какой-либо причине неспособные пересечь фосфолипидный бислой (например, из-за гидрофильных свойств, так как мембрана внутри гидрофобна и не пропускает гидрофильные вещества, или из-за крупных размеров), но необходимые для клетки, могут проникнуть сквозь мембрану через специальные белки-переносчики (транспортеры) и белки-каналы или путем эндоцитоза.
При пассивном транспорте вещества пересекают липидный бислой без затрат энергии по градиенту концентрации путем диффузии. Вариантом этого механизма является облегчённая диффузия, при которой веществу помогает пройти через мембрану какая-либо специфическая молекула. У этой молекулы может быть канал, пропускающий вещества только одного типа.
Активный транспорт требует затрат энергии, так как происходит против градиента концентрации. На мембране существуют специальные белки-насосы, в том числе АТФаза, которая активно вкачивает в клетку ионы калия (K+) и выкачивают из неё ионы натрия (Na+).
матричная — обеспечивает определенное взаиморасположение и ориентацию мембранных белков, их оптимальное взаимодействие.
механическая — обеспечивает автономность клетки, ее внутриклеточных структур, также соединение с другими клетками (в тканях). Большую роль в обеспечение механической функции имеют клеточные стенки, а у животных — межклеточное вещество.
энергетическая — при фотосинтезе в хлоропластах и клеточном дыхании в митохондриях в их мембранах действуют системы переноса энергии, в которых также участвуют белки;
рецепторная — некоторые белки, находящиеся в мембране, являются рецепторами (молекулами, при помощи которых клетка воспринимает те или иные сигналы).
Например, гормоны, циркулирующие в крови, действуют только на такие клетки-мишени, у которых есть соответствующие этим гормонам рецепторы. Нейромедиаторы (химические вещества, обеспечивающие проведение нервных импульсов) тоже связываются с особыми рецепторными белками клеток-мишеней.
ферментативная — мембранные белки нередко являются ферментами. Например, плазматические мембраны эпителиальных клеток кишечника содержат пищеварительные ферменты.
осуществление генерации и проведения биопотенциалов.
С помощью мембраны в клетке поддерживается постоянная концентрация ионов: концентрация иона К+ внутри клетки значительно выше, чем снаружи, а концентрация Na+ значительно ниже, что очень важно, так как это обеспечивает поддержание разности потенциалов на мембране и генерацию нервного импульса.
маркировка клетки — на мембране есть антигены, действующие как маркеры — «ярлыки», позволяющие опознать клетку. Это гликопротеины (то есть белки с присоединенными к ним разветвленными олигосахаридными боковыми цепями), играющие роль «антенн». Из-за бесчисленного множества конфигурации боковых цепей возможно сделать для каждого типа клеток свой особый маркер. С помощью маркеров клетки могут распознавать другие клетки и действовать согласованно с ними, например, при формировании органов и тканей. Это же позволяет иммунной системе распознавать чужеродные антигены.
Структура и состав биомембран
Мембраны состоят из липидов трёх классов: фосфолипиды, гликолипиды и холестерол. Фосфолипиды и гликолипиды (липиды с присоединёнными к ним углеводами) состоят из двух длинных гидрофобных углеводородных «хвостов», которые связаны с заряженной гидрофильной «головой». Холестерол придаёт мембране жёсткость, занимая свободное пространство между гидрофобными хвостами липидов и не позволяя им изгибаться. Поэтому мембраны с малым содержанием холестерола более гибкие, а с большим — более жёсткие и хрупкие. Также холестерол служит «стопором», препятствующим перемещению полярных молекул из клетки и в клетку. Важную часть мембраны составляют белки, пронизывающие её и отвечающие за разнообразные свойства мембран. Их состав и ориентация в разных мембранах различаются.
Клеточные мембраны часто асимметричны, то есть слои отличаются по составу липидов, переход отдельной молекулы из одного слоя в другой (так называемый флип-флоп) затруднён.
№28.Липосомы.
Липосомы — самопроизвольно образующиеся в смесях фосфолипидов с водой замкнутые пузырьки. Их стенка состоит из одного или нескольких бислоёв фосфолипидов (слоёв толщиной в две молекулы), в которые могут быть встроены другие вещества (например, белки). Внутри липосом содержится вода или раствор.
Диаметр липосом варьирует от 20 нм (моноламеллярные везикулы, стенка состоит из одного бислоя) до 10-50 мкм (мультиламеллярные везикулы, стенка состоит из десятков или сотен бислоёв).
Применение в медицине
С помощью липосом изучают воздействие на мембраны витаминов, гормонов, антибиотиков и других препаратов. Для ядовитых препаратов важным является точная их доставка к больному органу или ткани, минуя остальные части организма. Липосомы успешно используются, как носители лекарств, поскольку:
по химическому составу липосомы сходны с природными мембранами клеток;
липосомы универсальны, что позволяет переносить широкий спектр медицинских химических препаратов;
не вызывают аллергических реакций.
Липосомы широко применяются в экспериментальной онкологии. Однако есть ряд трудностей использования липосом в медицине. Во-первых, липосомы поглощаются клетками ретикуло-эндотелиальной системы, причём большее их количество находится в печени, селезенке, костном мозге, лимфатических узлах и кровотоке. Поэтому доставка лекарственных препаратов с помощью липосом в другие органы и части организма является более сложной задачей. Во-вторых, липопротеины, обмениваясь с липосомами липидами, способствуют разрушению липосом и вытеканию наружу их содержимого. Также стоит задача увеличения сроков хранения липосом после их приготовления.
№29.Ферменты.
Ферме́нты, или энзи́мы (от лат. fermentum, греч. ζύμη, ἔνζυμον — закваска) — обычно белковые молекулы или молекулы РНК (рибозимы) или их комплексы, ускоряющие (катализирующие) химические реакции в живых системах. Реагенты в реакции, катализируемой ферментами, называются субстратами, а получающиеся вещества — продуктами. Ферменты специфичны к субстратам (АТФаза катализирует расщепление только АТФ, а киназа фосфорилазы фосфорилирует только фосфорилазу).
Ферментативная активность может регулироваться активаторами и ингибиторами (активаторы — повышают, ингибиторы — понижают).
Белковые ферменты синтезируются на рибосомах, а РНК — в ядре.
Термины «фермент» и «энзим» давно используют как синонимы (первый в основном в русской и немецкой научной литературе, второй — в англо - и франкоязычной).
Наука о ферментах называется энзимологией, а не ферментологией (чтобы не смешивать корни слов латинского и греческого языков).Содержание [убрать]
1 История изучения
2 Функции ферментов
3 Классификация ферментов
4 Соглашения о наименовании ферментов
5 Кинетические исследования
6 Структура и механизм действия ферментов
6.1 Активный центр ферментов
История изучения
Термин фермент предложен в XVII веке химиком ван Гельмонтом при обсуждении механизмов пищеварения.
В кон. ХVIII — нач. XIX вв. уже было известно, что мясо переваривается желудочным соком, а крахмал превращается в сахар под действием слюны. Однако механизм этих явлений был неизвестен[1].
В XIX в. Луи Пастер, изучая превращение углеводов в этиловый спирт под действием дрожжей, пришёл к выводу, что этот процесс (брожение) катализируется некой жизненной силой, находящейся в дрожжевых клетках.
Более ста лет назад термины фермент и энзим отражали различные точки зрения в теоретическом споре Л. Пастера с одной стороны, и М. Бертло и Ю. Либиха — с другой, о природе спиртового брожения. Собственно ферментами (от лат. fermentum — закваска) называли «организованные ферменты» (то есть сами живые микроорганизмы), а термин энзим (от греч. ἐν- — в - и ζύμη — дрожжи, закваска) предложен в 1876 году В. Кюне для «неорганизованных ферментов», секретируемых клетками, например, в желудок (пепсин) или кишечник (трипсин, амилаза). Через два года после смерти Л. Пастера в 1897 году Э. Бухнер опубликовал работу «Спиртовое брожение без дрожжевых клеток», в которой экспериментально показал, что бесклеточный дрожжевой сок осуществляет спиртовое брожение так же, как и неразрушенные дрожжевые клетки. В 1907 году за эту работу он был удостоен Нобелевской премии. Впервые высокоочищенный кристаллический фермент (уреаза) был выделен в 1926 году Дж. Самнером. В течение последующих 10 лет было выделено еще несколько ферментов, и белковая природа ферментов была окончательно доказана.
№30.Свойства ферментов.
Ферменты называют катализаторами белковой природы, так как они в миллионы раз ускоряют течение отдельных химических реакций. Благодаря этой способности ферменты играют важнейшую роль в обмене веществ, во взаимодействии организма с внешней средой. Таким образом, первым свойством ферментов, отличающим их от химических катализаторов, является колоссальное ускорение течения реакций.
Вторым свойством ферментов является строгая специфичность их действия. Например, мальтаза разлагает мальтозу и не действует на близкий дисахарид сахарозу. Действие ферментов направлено на совершенно определенные химические связи.
Третьим свойством ферментов является их большая зависимость (лабильность) от ряда внешних воздействий — температуры, рН среды, окислительно-восстановительных условий, примесей некоторых веществ и др.
По степени чувствительности к температуре ферменты делят на две группы: термолабильные и термостабильные. Первые переносят кратковременное нагревание до 55— 65 °С, при более высокой температуре они инактивируются. Вторые переносят нагревание до температуры ~75°С.
Примером высокоустойчивого к нагреванию внеклеточного фермента является а-амилаза.
Интересен вопрос о происхождении ферментов. По представлению некоторых ученых ферменты возникли на Земле одновременно или даже раньше, чем первичные формы жизни. По-видимому, предшественниками их являлись неорганические катализаторы или органические соединения с низким молекулярным весом. Однако они обладали малой специфичностью и слабым каталитическим действием и по существу еще не являлись ферментами.
Соединение таких катализаторов с белками положило начало возникновению ферментов. В дальнейшем первичные ферменты подвергались естественному отбору и сохранялись только те, которые улучшали жизненные процессы организмов.
№31.Химическая природа ферментов.
Изучение механизма химической реакции, катализируемой ферментом наряду с определением промежуточных и конечных продуктов на разных стадиях реакции подразумевает точное знание геометрии третичной структуры фермента, природы функциональных групп его молекулы, обеспечивающих специфичность действия и высокую каталитическую активность на данный субстрат, а также химической природы участка (участков) молекулы фермента, который обеспечивает высокую скорость каталитической реакции. Обычно молекулы субстрата, участвующие в ферментативных реакциях, по сравнению с молекулами ферментов имеют относительно небольшие размеры. Таким образом, при образовании фермент-субстратных комплексов в непосредственное химическое взаимодействие вступают лишь ограниченные фрагменты аминокислотной последовательности полипептидной цепи — «активный центр» — уникальная комбинация остатков аминокислот в молекуле фермента, обеспечивающая непосредственное взаимодействие с молекулой субстрата и прямое участие в акте катализа[8].
В активном центре условно выделяют[8]:
каталитический центр — непосредственно химически взаимодействующий с субстратом;
связывающий центр (контактная или «якорная» площадка) — обеспецивающий специфическое сродство к субстрату и формирование комплекса фермент-субстрат.
Чтобы катализировать реакцию, фермент должен связаться с одним или несколькими субстратами. Белковая цепь фермента сворачивается таким образом, что на поверхности глобулы образуется щель, или впадина, где связываются субстраты. Эта область называется сайтом связывания субстрата. Обычно он совпадает с активным центром фермента или находится вблизи него. Некоторые ферменты содержат также сайты связывания кофакторов или ионов металлов.
Фермент, соединяясь с субстратом:
очищает субстрат от водяной «шубы»
располагает реагирующие молекулы субстратов в пространстве нужным для протекания реакции образом
подготавливает к реакции (например, поляризует) молекулы субстратов.
Обычно присоединение фермента к субстрату происходит за счет ионных или водородных связей, редко — за счет ковалентных. В конце реакции её продукт (или продукты) отделяются от фермента.
В результате фермент снижает энергию активации реакции. Это происходит потому, что в присутствии фермента реакция идет по другому пути (фактически происходит другая реакция), например:
В отсутствие фермента:
А+В = АВ
В присутствии фермента:
А+Ф = АФ
АФ+В = АВФ
АВФ = АВ+Ф
где А, В — субстраты, АВ — продукт реакции, Ф — фермент.
Ферменты не могут самостоятельно обеспечивать энергией эндергонические реакции (для протекания которых требуется энергия). Поэтому ферменты, осуществляющие такие реакции, сопрягают их с экзергоническими реакциями, идущими с выделением большего количества энергии. Например, реакции синтеза биополимеров часто сопрягаются с реакцией гидролиза АТФ.
Для активных центров некоторых ферментов характерно явление кооперативности.
№32.Кофакторы и коферменты.
Некоторые ферменты выполняют каталитическую функцию сами по себе, безо всяких дополнительных компонентов. Однако есть ферменты, которым для осуществления катализа необходимы компоненты небелковой природы. Кофакторы могут быть как неорганическими молекулами (ионы металлов, железо-серные кластеры и др.), так и органическими (например, флавин или гем). Органические кофакторы, прочно связанные с ферментом, называют также простетическими группами. Кофакторы органической природы, способные отделяться от фермента, называют коферментами.
Фермент, который требует наличия кофактора для проявления каталитической активности, но не связан с ним, называется апо-фермент. Апо-фермент в комплексе с кофактором носит название холо-фермента. Большинство кофакторов связано с ферментом нековалентными, но довольно прочными взаимодействиями. Есть и такие простетические группы, которые связаны с ферментом ковалентно, например, тиаминпирофосфат в пируватдегидрогеназе.
Коферменты, или коэнзимы — малые молекулы небелковой природы, специфически соединяющиеся с соответствующими белками, называемыми апоферментами, и играющие роль активного центра или простетической группы молекулы фермента.
Комплекс кофермента и апофермента образует целостную, биологически активную молекулу фермента, называемую холоферментом
Роль коферментов нередко играют витамины или их метаболиты (чаще всего — фосфорилированные формы витаминов группы B). Например, коферментом фермента карбоксилазы является тиаминпирофосфат, коферментом многих аминотрансфераз — пиридоксаль-6-фосфат.
В металлоферментах роль, аналогичную роли коферментов, могут исполнять катионы металлов, однако коферментами их обычно не называют.
№34,35.Константа Михаэлиса.
Одним из характерных проявлений жизни является удивительная способность живых организмов кинетически регулировать химические реакции, подавляя стремление к достижению термодинамического равновесия. Ферментативная кинетика занимается исследованием закономерностей влияния химической природы реагирующих веществ (ферментов, субстратов) и условий их взаимодействия (концентрация, рН среды, температуры, присутствие активаторов или ингибиторов) на скорость ферментативной реакции. Главной целью изучения кинетики ферментативных реакций является получение информации, которая может способствовать выяснению молекулярного механизма действия фермента.
Общие принципы кинетики химических реакций применимы и к ферментативным реакциям. Известно, что любая химическая реакция характеризуется константой термодинамического равновесия. Она выражает состояние химического равновесия, достигаемого системой, и обозначается Кр. Так, для реакции:
![]()
константа равновесия равна произведению концентраций образующихся веществ, деленному на произведение концентрации исходных веществ. Значение константы равновесия обычно находят из соотношения констант скоростей прямой (k+1) и обратной (k– 1 ) реакций, т. е. Кp = k+1/k–1. В состоянии равновесия скорость прямой реакции: v+1 = k + 1[ А ] • [ B ] равна скорости обратной реакции: v–1 = k – 1 [ С ] • [ D ] , т. е. v+1 = v–1 соответственно k+1[А]•[B] = k–1[С]•[D], или
![]()
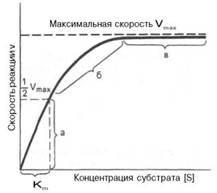
Рис. 4.12. Теоретический график зависимости скорости ферментативной реакции от концентрации субстрата при постоянной концентрации фермента.
а - реакция первого порядка (при [ S ] < Кm скорость реакции пропорциональна концентрации субстрата); б - реакция смешанного порядка; в - реакция нулевого порядка, когда v = Vmaxи скорость реакции не зависит от концентрации субстрата.
Таким образом, константа равновесия равна отношению констант скоростей прямой и обратной реакций. Величину, обратную константе равновесия, принято называть субстратной константой, или, в случае ферментативной реакции, константой диссоциации фермент–субстратного комплекса, и обозначать символом KS. Так, в реакции
![]()
т. е. KSравна отношению произведения концентрации фермента и субстрата к концентрации фермент-субстратного комплекса или отношению констант скоростей обратной и прямой реакций. Следует отметить, что константа KSзависит от химической природы субстрата и фермента и определяет степень их сродства. Чем ниже значение KS, тем выше сродство фермента к субстрату.
При изучении кинетики ферментативных реакций следует учитывать одну важную особенность этих реакций (не свойственную обычным химическим реакциям), связанную с явлением насыщения фермента субстратом. При низкой концентрации субстрата зависимость скорости реакции от концентрации субстрата (рис. 4.12) является почти линейной и подчиняется кинетике первого порядка. Это означает, что скорость реакции S —> Р прямо пропорциональна концентрации субстрата S и в любой момент времени t определяется следующим кинетическим уравнением:
![]()
где [S] – молярная концентрация субстрата S; –d[S]/dt – скорость убыли субстрата; k' – константа скорости реакции, которая в данном случае имеет размерность, обратную единице времени (мин–1 или с–1).
При высокой концентрации субстрата скорость реакции максимальна, становится постоянной и не зависящей от концентрации субстрата [ S ] . В этом случае реакция подчиняется кинетике нулевого порядка v = k" (при полном насыщении фермента субстратом) и целиком определяется концентрацией фермента. Различают, кроме того, реакции второго порядка, скорость которых пропорциональна произведению концентраций двух реагирующих веществ. В определенных условиях при нарушении пропорциональности говорят иногда о реакциях смешанного порядка (см. рис. 4.12).
Изучая явление насыщения, Л. Михаэлис и М. Ментен разработали общую теорию ферментативной кинетики. Они исходили из предположения, что ферментативный процесс протекает в виде следующей химической реакции:
![]()
т. е. фермент Е вступает во взаимодействие с субстратом S с образованием промежуточного комплекса ES, который далее распадается на свободный фермент и продукт реакции Р. Математическая обработка на основе закона действующих масс дала возможность вывести уравнение, названное в честь авторов уравнением Михаэлиса–Ментен, выражающее количественное соотношение между концентрацией субстрата и скоростью ферментативной реакции:
![]()
где v – наблюдаемая скорость реакции при данной концентрации субстрата [S]; KS– константа диссоциации фермент-субстратного комплекса, моль/л; Vmax– максимальная скорость реакции при полном насыщении фермента субстратом.
Из уравнения Михаэлиса–Ментен следует, что при высокой концентрации субстрата и низком значении KSскорость реакции является максимальной, т. е. v = Vmax(реакция нулевого порядка, см. рис. 4.12). При низкой концентрации субстрата, напротив, скорость реакции оказывается пропорциональной концентрации субстрата в каждый данный момент (реакция первого порядка).
Следует указать, что уравнение Михаэлиса–Ментен в его классическом виде не учитывает влияние на скорость ферментативного процесса продуктов реакции, например в реакции
![]()
и носит несколько ограниченный характер. Поэтому были предприняты попытки усовершенствовать его. Так, было предложено уравнение Бриггса-Холдейна:
![]()
где Кm представляет собой константу Михаэлиса, являющуюся экспериментально определяемой величиной. Она может быть представлена следующим уравнением:
![]()
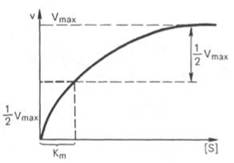
Рис. 4.13. Кривая уравнения Михаэли-са-Ментен: гиперболическая зависимость начальных скоростей катализируемой ферментом реакции от концентрации субстрата.
В числителе представлены константы скоростей распада комплекса ES в двух направлениях (в сторону исходных Е и S и в сторону конечных продуктов реакции Е и Р). Отношение k–1/ k+1представляет собой константу диссоциации ферментсубстратного комплекса KS, тогда:
![]()
Отсюда вытекает важное следствие: константа Михаэлиса всегда больше константы диссоциации фермент-субстратного комплекса KSна величину
k+2/k+1.
Для определения численного значения Кm обычно находят ту концентрацию субстрата, при которой скорость ферментативной реакции v составляет половину от максимальной Vmax, т. е. если v = 1/2 Vmaх. Подставляя значение v в уравнение Бриггса–Холдейна, получаем:
![]()
![]()
Таким образом, константа Михаэлиса численно равна концентрации субстрата (моль/л), при которой скорость данной ферментативной реакции составляет половину от максимальной.
Определение величины Кm имеет важное значение при выяснении механизма действия эффекторов на активность ферментов и т. д. Константу Михаэлиса можно вычислить по графику (рис. 4.13). Отрезок на абсциссе, соответствующий скорости, равной половине максимальной, будет представлять собой Кm.
Пользоваться графиком, построенным в прямых координатах зависимости начальной скорости реакции v0 от начальной концентрации субстрата [S0], неудобно, поскольку максимальная скорость Vmaxявляется в данном случае асимптотической величиной и определяется недостаточно точно.
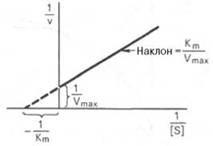
Рис. 4.14. График Лайнуивера-Бэрка.
Для более удобного графического представления экспериментальных данных Г. Лайнуивер и Д. Бэрк преобразовали уравнение Бриггса–Хол-дейна по методу двойных обратных величин исходя из того принципа, что если существует равенство между двумя какими-либо величинами, то и обратные величины также будут равны. В частности, если
![]()
![]()
которое получило название уравнения Лайнуивера–Бэрка. Это уравнение прямой линии: у = ах + b. Если теперь в соответствии с этим уравнением построить график в координатах 1/v (y) от l/[S] (x), то получим прямую линию (рис. 4.14), тангенс угла наклона который будет равен величине Km/Vmax; отрезок, отсекаемый прямой от оси ординат, представляет собой l/Vmax(обратная величина максимальной скорости). Если продолжить прямую линию за ось ординат, тогда на абсциссе отсекается отрезок, соответствующий обратной величине константы Михаэлиса – 1/Кm (см. рис. 4.14). Таким образом, величину Кm можно вычислить из данных наклона прямой и длины отрезка, отсекаемого от оси ординат, или из длины отрезка, отсекаемого от оси абсцисс в области отрицательных значений.
Следует подчеркнуть, что значения Vmax, как и величину Кm, более точно, чем по графику, построенному в прямых координатах, можно определить по графику, построенному по методу двойных обратных величин. Поэтому данный метод нашел широкое применение в современной энзимологии. Предложены также аналогичные графические способы определения Кm и Vmaxв координатах зависимости v от v/[S] и [S]/v от [S].
Следует отметить некоторые ограничения применения уравнения Ми-хаэлиса–Ментен, обусловленные множественными формами ферментов и аллостерической природой фермента. В этом случае график зависимости начальной скорости реакции от концентрации субстрата (кинетическая
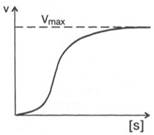
Рис. 4.15. Сигмоидная кинетическая кривая насыщения субстратом.
кривая) имеет не гиперболическую форму, а сигмоидный характер (рис. 4.15) наподобие кривой насыщения гемоглобина кислородом. Это означает, что связывание одной молекулы субстрата в одном каталитическом центре повышает связывание субстрата с другим центром, т. е. имеет место кооперативное взаимодействие, как и в случае присоединения кислорода к 4 субъединицам гемоглобина. Для оценки концентрации субстрата, при которой скорость реакции составляет половину максимальной, в условиях сигмоидного характера кинетической кривой обычно применяют преобразованное уравнение Хилла:
![]()
где К' – константа ассоциации; n – число субстратсвязывающих центров.
№36,37.Активаторы и ингибиторы ферментов.
Вещества, которые оказывают влияние на активность ферментов, называют эффекторами. Это могут быть ингибиторы – соединения, тормозящие каталитический процесс, или активаторы – вещества, которые этот процесс ускоряют. Учение об ингибиторах ферментов имеет большое теоретическое и практическое значение для фармакологии и токсикологии. Многие лекарственные препараты являются ингибиторами ферментов. Например, ингибиторы амилаз успешно применяются для лечения заболеваний, связанных с повышенной активностью этих ферментов – диабета, ожирения, кариеса. Используемые в военном деле нервно-паралитические газы представляют собой специфические ингибиторы ферментов. В научных исследованиях специфические ингибиторы используются для изучения механизма действия ферментов, строения их активного центра. Например, многие из промежуточных продуктов гликолиза и дрожжевого брожения были открыты благодаря использованию ингибиторов, блокирующих последовательные стадии процесса. В результате такого блокирования соответствующие промежуточные продукты накапливались в количествах, достаточных для их выделения и идентификации.
По типу действия ингибиторы можно разделить на обратимые и необратимые. Удаление обратимых ингибиторов из системы (диализом, гельфильтрацией и др.) восстанавливает каталитическую активность фермента.
Обратимо действуют эффекторы:
1. Близкие аналоги субстрата, которые связываются активным центром фермента, но
не подвергаются превращению. Занимая активный центр, они препятствуют связыванию истинного субстрата, конкурируя с ним, и поэтому называются конкурентными ингибиторами.
2. Кофакторы ферментов, без которых апофермент вообще не обладает активностью.
Постепенное добавление их приводит к появлению активности, которая затем повышается до определенного предела, соответствующего полному насыщению.
3. Вещества, которые взаимодействуют с дополнительными, регуляторными центрами, несовпадающими с активным центром. Тем не менее, это взаимодействие изменяет конформацию в районе активного центра и влияет на кинетику ферментативного процесса. Такие соединения называются аллостерическими эффекторами. Они имеют важное биологическое значение, так как с их помощью осуществляется один из механизмов регуляции каталитической активности.
Необратимую инактивацию вызывают соединения (найденные в живой природе или полученные путем синтеза), которые вступают в химическую реакцию с участком фермента, важным для проявления каталитической активности. Такие соединения, специфически реагирующие с определенными группами в молекулах ферментов (групп-специфические реагенты), используют для идентификации функциональных групп активного центра (метод химической модификации).
С этой целью широко используются соединения, блокирующие SH-группы (иодацетамид, n-хлормеркурибензоат и др.), окисляющие остатки триптофана в кислой среде (N-бромсукцинимид), ацетилирующие остатки тирозина (N-ацетилимидазол), связывающие металлы (азид натрия) и т. д.
№38.Механизм действия ферментов.
Структура и функции ферментов, а также механизм их действия почти ежегодно подробно обсуждаются на многих международных симпозиумах и конгрессах. Важное место отводится рассмотрению структуры всей молекулы фермента и ее активных центров, молекулярному механизму действия различных типов ферментов, общей теории энзиматического катализа. Тем не менее до сих пор нет полной ясности по двум кардинальным проблемам энзимологии: чем вызваны специфичность действия и высокая каталитическая эффективность ферментов?
До установления химической природы ферментов гипотезы о механизме их действия опирались на исследования кинетики и модельные опыты химического гомогенного катализа. Повышение скорости химических реакций под действием ферментов объясняли следующим: а) активированием субстрата в результате образования адсорбционных или молекулярных, обратимо диссоциирующих фермент-субстратных комплексов; б) цепным механизмом реакций с участием радикалов или возбужденных молекул. Оказалось, что цепные механизмы реакции не играют существенной роли в биологическом катализе. После установления химической природы ферментов подтвердилось представление, выдвинутое более 80 лет назад В. Анри, Л. Михаэлисом и М. Ментен, о том, что при энзиматическом катализе фермент Е соединяется (в принципе обратимо) со своим субстратом S, образуя нестойкий промежуточный фермент-субстратный комплекс ES, который в конце реакции распадается с освобождением фермента и продуктов реакции Р. Благодаря высокому сродству связывания и образованию ES-комплекса резко возрастает число молекул субстрата, вступающих в реакции. Эти представления легли в основу теории «ключа-замка» Э. Фишера, которую иногда называют теорией «жесткой матрицы». Таким образом, жесткая структура активного центра оказывается комплементарной молекулярной структуре субстрата, обеспечивая тем самым высокую специфичность фермента.
Л. Михаэлис не только постулировал образование промежуточного фермент-субстратного ES-комплекса, но и рассчитал влияние концентрации субстрата на скорость реакции. В процессе реакции различают несколько стадий: присоединение молекулы субстрата к ферменту, преобразование первичного промежуточного соединения в один или несколько последовательных (переходных) комплексов и протекающее в одну или несколько стадий отделение конечных продуктов реакции от фермента.
Фермент вступает во взаимодействие с субстратом на очень короткий период, поэтому долгое время не удавалось показать образование такого комплекса. Прямые доказательства существования фермент-субстратного комплекса были получены в лабораториях Д. Кейлина и Б. Чанса. В настоящее время экспериментальные и математические методы кинетики, термодинамики и статической механики химических реакций позволяют
определить для ряда ферментативных реакций кинетические и термодинамические показатели, в частности константы диссоциации промежуточных фермент-субстратных комплексов, константы скорости и равновесия их образования.
В образовании фермент-субстратных комплексов участвуют водородные связи, электростатические и гидрофобные взаимодействия, а в ряде случаев также ковалентные, координационные связи (рис. 4.9). Информация о природе связей между субстратом и связывающим участком активного центра фермента может быть получена методами ЭПР и ЯМР, а также методами УФ - и ИК-спектроскопии.
Для каталитической активности фермента существенное значение имеет пространственная структура, в которой жесткие участки α-спиралей чередуются с гибкими, эластичными линейными отрезками, обеспечивающими динамические изменения белковой молекулы фермента. Этим изме-неням придается большое значение в некоторых теориях ферментативного катализа. Так, в противоположность модели Э. Фишера «ключ-замок» Д. Кошлендом была разработана теория «индуцированного соответствия», допускающая высокую конформационную лабильность молекулы белка-фермента и гибкость и подвижность активного центра. Эта теория была основана на весьма убедительных экспериментах, свидетельствующих о том, что субстрат индуцирует конформационные изменения молекулы фермента таким образом, что активный центр принимает необходимую для связывания субстрата пространственную ориентацию. Иными словами, фермент только в присутствии (точнее, в момент присоединения) субстрата будет находиться в активной (напряженной) Т-форме в отличие от неактивной R-формы (рис. 4.10). На рис. 4.10 видно, что присоединение субстрата S к ферменту Е, вызывая соответствующие изменения конформации активного центра, в одних случаях приводит к образованию активного комплекса, в других – неактивного комплекса вследствие нарушения пространственного расположения функциональных групп активного центра в промежуточном комплексе. Получены экспериментальные доказательства нового положения о том, что постулированное Д. Кошлендом «индуцированное соответствие» субстрата и фермента создается не обязательно изменениями конформации белковой молекулы, но также геометрической и электронно-топографической перестройкой молекулы субстрата.
В каталитическом процессе существенное значение имеют точное соответствие между ферментом и субстратом, а также термодинамические и каталитические преимущества подобного соответствия. Гипотеза «индуцированного соответствия» предполагает существование между ферментом и субстратом не только пространственной или геометрической компле-ментарности, но и электростатического соответствия, обусловленного спариванием противоположно заряженных групп субстрата и активного центра фермента. Точное соответствие обеспечивает образование эффективного комплекса между субстратом и ферментом.

|
Из за большого объема этот материал размещен на нескольких страницах:
1 2 3 4 |
