
Партнерка на США и Канаду по недвижимости, выплаты в крипто
- 30% recurring commission
- Выплаты в USDT
- Вывод каждую неделю
- Комиссия до 5 лет за каждого referral
УДК 544.16; 544.31
ИССЛЕДОВАНИЕ СТЕХИОМЕТРИЧЕСКИХ МОНОМЕРОВ КЛАСТЕРА ОКСИДА АЛЮМИНИЯ МЕТОДАМИ КВАНТОВОЙ ХИМИИ
Федеральное государственное унитарное предприятие «Центральный институт авиационного моторостроения имени »
Введение
В последние годы значительный интерес проявляется к исследованиям кинетических процессов формирования наночастиц в низкотемпературной пылевой плазме [1], образующейся при горении углеводородных и металлизированных топлив [2]. С повышением производительности вычислительной техники стали возможны квантовохимические расчёты поверхностей потенциальной энергии кластеров, состоящих из нескольких мономеров, и их физических свойств [3].
Теоретический анализ структуры межмолекулярных комплексов и кластеров методами квантовой химии является одним из важных направлений в физике кластеров [4, 5]. Обычно под кластером понимают объект диаметром до нескольких нанометров, содержащий от двух до нескольких сотен мономеров (атомов или молекул).
Исследование свойств кластера предполагает рассмотрение самой элементарной структуры — кластерообразующей формульной единицы.
Поскольку кластеры представляют собой промежуточные образования между молекулярным и конденсированным состояниями вещества, результаты исследований в рамках кластерных моделей относятся к самым основам теории строения вещества. С точки зрения приложения квантовохимических методик надёжный расчёт свойств межмолекулярных комплексов и кластеров оказывается достаточно сложной задачей, так как окончательные оценки энергии и оптимизация геометрических параметров должны быть выполнены с учётом эффектов электронной корреляции [3].
Цель работы
Работа посвящена нахождению структуры и свойств кластеров оксида алюминия (Al2O3)n, n=1. Основной задачей являлось нахождение молекулярных постоянных и термодинамических функций мономеров Al2O3 через расчёт их геометрических и энергетических параметров методами квантовой химии. Требовалось определить ту конфигурацию атомов, которой соответствует наименьшая энергия основного состояния на поверхности потенциальной энергии системы. Для этого была проведена оптимизация структуры кластера — определение оптимальных геометрических параметров.
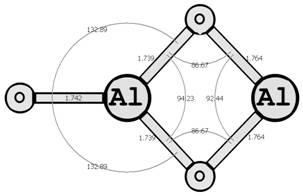
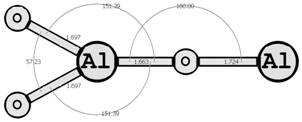
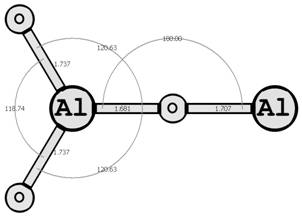
Предыдущие исследования выявили два предпочтительных изомера разной мультиплетности с наинизшей энергией и группой симметрии: D∞h (1Σg+) [6, 7] и C2v (3B2) [8, 9] (здесь и далее цифра в верхнем индексе обозначает мультиплетность молекулярной системы: единица — синглетное состояние, тройка — триплетное). Были рассмотрены стехиометрические соединения алюминия с кислородом Al2O3 (рис. 1): линейный D∞h (1Σg+) и плоский Cs (3A1′), бипирамидальные D3h (1A1′) и D3h (3A2′) (исследованные ранее в [10, 11]), а также по две конфигурации плоских C2v (1A1) и C2v (3B2) и Y-образных [12, 13] (игрекобразных) C2v (1A1′) и C2v (3A1′).
Для этих восьми структур найдены длины межатомных расстояний (в Å), величины валентных и двугранных углов, частоты нормальных колебаний νi, моменты инерции Jx, Jy, Jz (вращательные постоянные Bx, By, Bz), группы симметрии Шёнфлиса, вращательные числа симметрии σ и полные энергии основного состояния системы E(Al2O3).

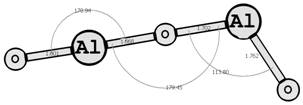
D∞h (1Σg+) Cs (3A1′)
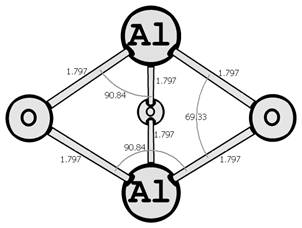
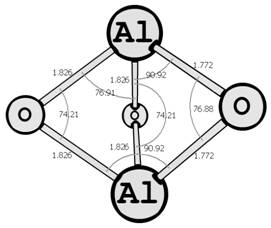
D3h (1A1′) D3h (3A2′)
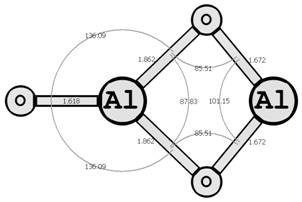
C2v (1A1) C2v (3B2)
C2v (1A1′) C2v (3A1′)
Рис. 1. Возможные конфигурации мономеров (в левом столбце расположены синглеты, в правом — триплеты) (длины связей измерены в ангстремах, углы — в градусах).
Методология
Была проведена оптимизация равновесной геометрии кластеров, то есть определялась структура, соответствующая локальному минимуму потенциальной энергии молекулярной системы на поверхности потенциальной энергии. Нахождение молекулярных волновых функций осуществлено при решении уравнений самосогласованного поля (SCF) неограниченным методом Хартри — Фока (UHF) [3] и на основе теории функционала электронной плотности (DFT) с использованием трёхпараметрического обменного функционала Беке [14] в комбинации с корреляционным функционалом Ли — Янга — Парра [15] (B3LYP) для учёта энергии электронной корреляции. Далее вычислялся гессиан потенциальной энергии и частоты нормальных колебаний. Расчёт частот нормальных колебаний позволил проверить, действительно ли получено наилучшее взаимное расположение атомов, то есть отвечает ли найденная пространственная конфигурация истинной точке локального минимума на поверхности потенциальной энергии. Квантовохимический расчёт был проведён в поляризационном трёхэкспоненциальном валентно-расщеплённом базисном наборе Попла 6-311++G(3df), включающем диффузные функции [16], с помощью пакета программ Firefly 7.1.G [17].
Результаты расчётов
Энергия атомизации мономеров была вычислена следующим образом:
Eатом.=2E(2Al)+3E(3O)–E(Al2O3),
где E(2Al)=–242,3881791 а. е., E(3O)=–75,0932238 а. е., к которой была добавлена энергия нулевых колебаний ![]() (f — количество колебательных частот), чтобы получить Eа=Eатом.–E0.
(f — количество колебательных частот), чтобы получить Eа=Eатом.–E0.
Табл. 1. Молекулярные постоянные (здесь [Eатом.]=[E0]=[Eа]=кДж/моль, [Bx]=[By]=[Bz]=К, причём 1 а. е.≈2633 кДж/моль).
Изомер | σ | Bx | By | Bz | E(Al2O3), а. е. | Eатом. | E0 | Eа |
C2v (3B2) | 2 | 0,47 | 0,13 | 0,1 | –710,7803347 | 1907 | 29 | 1878 |
D∞h (1Σg+) | 2 | 0,05 | –710,7678085 | 1874 | 29 | 1845 | ||
C2v (1A1) | 2 | 0,46 | 0,13 | 0,1 | –710,750322 | 1828 | 30 | 1798 |
C2v (1A1′) | 2 | 1,15 | 0,07 | 0,06 | –710,7256962 | 1763 | 27 | 1736 |
C2v (3A1′) | 2 | 0,34 | 0,08 | 0,07 | –710,7195156 | 1747 | 23 | 1724 |
D3h (3A2′) | 6 | 0,25 | 0,23 | 0,2 | –710,7171298 | 1740 | 28 | 1712 |
Cs (3A1′) | 1 | 1 | 0,06 | 0,05 | –710,7083152 | 1717 | 25 | 1692 |
D3h (1A1′) | 6 | 0,23 | 0,22 | 0,22 | –710,6996869 | 1695 | 29 | 1666 |
Видно, что значение энергии атомизации с учётом нулевых колебаний Eа изомера C2v(3B2) с наименьшей энергией основного состояния неплохо согласуется с табличным значением [18].
Вычисление термодинамических функций
Статистическая сумма по состояниям молекулы равна произведению поступательной, вращательной, колебательной и электронной статистических сумм:
Q=Qпост.Qвращ.Qкол.Qэл.,
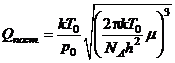 ,
, 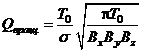 ,
, 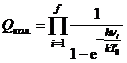 .
.
Использовано приближение жёсткого ротатора и гармонического осциллятора, причём выбраны стандартные условия: T0=298,15 К, p0=101325 Па. Здесь молярная масса μ(Al2O3)=0,102 кг/моль; k — постоянная Больцмана, NA — постоянная Авогадро, h — постоянная Планка.
Линейный D∞h (1Σg+) мономер имеет десять частот, так как он обладает двумя, а не тремя вращательными степенями свободы, и, в отличие от других изомеров, его общее количество внутренних степеней свободы равно 3∙5–(3+2)=10, а не девяти. Для линейной молекулы ![]() , где
, где ![]() .
.
Табл. 2. Частоты нормальных колебаний (здесь [ν]=ТГц) (число в скобках означает кратность вырождения колебательной моды).
Изомер | Qэл. | ν1 | ν2 | ν3 | ν4 | ν5 | ν6 | ν7 | ν8 | ν9 |
C2v (3B2) | 3 | 4,27 | 5,32 | 10,58 | 13,61 | 19,22 | 20,47 | 22,5 | 23,23 | 27,78 |
D∞h (1Σg+) | 1 | 1,5 (2) | 6,1 (2) | 8,54 (2) | 13,15 | 27,98 | 34,29 | 36,76 | ||
C2v (1A1) | 1 | 4,72 | 5,92 | 10,25 | 13,68 | 14,73 | 15,88 | 24,68 | 25,84 | 29,61 |
C2v (1A1′) | 1 | 1,82 | 2,02 | 7,22 | 7,56 | 12,25 | 19,03 | 22,94 | 27,06 | 34,12 |
C2v (3A1′) | 3 | 1,53 | 2,4 | 5,52 | 6,91 | 8,51 | 14,29 | 22,19 | 25,32 | 30,33 |
D3h (3A2′) | 3 | 9,84 | 9,92 | 10,89 | 11,9 | 15,09 | 16,9 | 18,58 | 21,48 | 23,25 |
Cs (3A1′) | 3 | 1,35 | 1,98 | 5,38 | 7,75 | 8,14 | 14,08 | 22,18 | 29,34 | 35,86 |
D3h (1A1′) | 1 | 10,33 | 10,38 | 12,98 | 13 | 17,57 | 18,79 | 18,85 | 21,2 | 23,8 |
Найдём молярную теплоёмкость при постоянном давлении cp, энтропию S, энтальпию H, энергию Гиббса G=H–TS в соответствии с [18]:
![]() , Дж/(моль·К),
, Дж/(моль·К),
![]() , Дж/(моль·К),
, Дж/(моль·К),
![]() , Дж/(моль),
, Дж/(моль),
где H298=Eэл.+E0+Eколеб.+Eпост.+Eвращ.+RT0— полная энтальпия, включающая тепловую энергию поступательного движения, Eпост.=3/2·RT0 (как и для атомов), Eвращ.=3/2·RT0 для нелинейных молекул и Eвращ.=2/2·RT0 для линейных молекул (2 степени свободы), 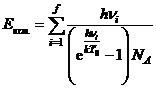 , R=kNA — универсальная газовая постоянная.
, R=kNA — универсальная газовая постоянная.
Табл. 3. Термодинамические величины (значения взяты при T0=298,15 К) (здесь [cp]=[S298]=Дж/(моль·К), [Eкол.]=[H298]=[ΔfH298]=[ΔfG298]=кДж/моль.
Изомер | Eколебат. | cp | S298 | H298–Eэл. | ΔfH298 | ΔfG298 |
C2v (3B2) | 6 | 77 | 315 | 61 | –416 | –413 |
D∞h (1Σg+) | 11 | 85 | 315 | 67 | –376 | –373 |
C2v (1A1) | 6 | 77 | 305 | 60 | –337 | –331 |
C2v (1A1′) | 8 | 81 | 329 | 62 | –271 | –270 |
C2v (3A1′) | 9 | 85 | 345 | 60 | –256 | –250 |
D3h (3A2′) | 6 | 79 | 299 | 57 | –253 | –244 |
Cs (3A1′) | 9 | 84 | 352 | 62 | –225 | –217 |
D3h (1A1′) | 5 | 77 | 286 | 58 | –206 | –194 |
Энтальпии образования ΔfH298(Al)=325,5 кДж/моль, ΔfH298(O)=249 кДж/моль из [19], энтропии S298(Al)=165 Дж/(моль·К), S298(O)=161 Дж/(моль·К) позволяют найти энтальпию образования ΔfH298 и энергию Гиббса образования ΔfG298:
2·{ΔfH298(Al)–(E(2Al)+Eпост.+RT0)}+3·{ΔfH298(O)–(E(3O)+Eпост.+RT0)}+
+E(Al2O3)+H298(Al2O3)=ΔfH298(Al2O3),
2·{ΔfH298(Al)–(E(2Al)+Eпост.+RT0)}–T0S298(Al)+3·{ΔfH298(O)–(E(3O)+Eпост.+RT0)}–
–T0S298(O)+E(Al2O3)+H298(Al2O3)+T0S298(Al2O3)=ΔfG298(Al2O3).
Заключение
Итак, для всех возможных изомеров были определены пространственные структуры и такие характеристики, как электронные энергии основного состояния, нулевых колебаний, атомизации, молярная теплоёмкость при постоянном давлении, энтропия, энтальпия и энергия Гиббса образования оксида алюминия.
Квантовохимические расчёты показали, что наименьшей энергией среди всех возможных изомеров различной мультиплетности обладает триплетный мономер группы симметрии C2v Al2O3(3B2). Значение, полученное для его энергии атомизации, неплохо согласуется с экспериментальными данными. В то же время линейный синглетный изомер D∞h (1Σg+) лежит на 0,34 эВ выше по энергии. Этот факт необходимо учитывать при расчёте термодинамических функций газообразного оксида алюминия.
Значительный разброс термодинамических характеристик мономеров (при одной и той же температуре) предполагает введение распределения их значений, чтобы определить процентный состав каждого изомера в совокупности всех мономеров при указанной температуре. Нахождение средних значений термодинамических функций при заданных температурах позволит получить усреднённые теплофизические величины.
Поскольку в данной работе исследовались всего лишь четыре конфигурации Al2O3 с двумя значениями мультиплетности, следует обратить внимание на другие возможные изомеры: с иным расположением атомов (например, кольцеобразным), с большей мультиплетностью (пять и выше).
Список литературы
[1] , , . Механизмы формирования кластеров и наночастиц в плазме, образующейся при горении углеводородных и металлизированных топлив // Неравновесные физико-химические процессы в газовых потоках и новые принципы организации горения. — М.: ТОРУС ПРЕСС, 2011. С. 305—326.
[2] , , . Кинетика окисления углеводородных топлив, содержащих наночастицы алюминия // Неравновесные физико-химические процессы в газовых потоках и новые принципы организации горения. — М.: ТОРУС ПРЕСС, 2011. — С. 131—159.
[3] A. В. Немухин, , . Молекулярное моделирование с программой PC GAMESS: от двухатомных молекул до ферментов. — Вестник Московского Университета. Сер. 2. Химия. — Т. 45. — № 2. С. 75—102; 2004.
[4] A. B. C. Patzer, Ch. Chang, E. Sedlmayr, D. Sülzle. A density functional study of small AlxOy (x, y=1—4) clusters and their thermodynamic properties. — Eur. Phys. J. D. — Vol. 32, pp. 329—337; 2005.
[5] , . Исследование стехиометрических кластеров оксида алюминия методами квантовой химии. — Труды 54-й научной конференции МФТИ «Проблемы фундаментальных и прикладных естественных и технических наук в современном информационном обществе. Аэромеханика и летательная техника». — М.: МФТИ, 2011. С. 17—19.
[6] A. V. Nemukhin, F. Weinhold. Structures of the aluminum oxides studied by ab initio methods with natural bond orbital analysis. — J. Chem. Phys. — Vol. 97, № 5, pp. 3420—3430; 1992.
[7] M. T. Swihart, L. Catoire. Thermochemistry of aluminum species for combustion modeling from ab initio molecular orbital calculations. — Combust. Flame. — Vol. 121, pp. 210—222; 2000.
[8] E. F. Archibong, A. St-Amant. On the structure of Al2O3 and photoelectron spectra of Al2O2– and Al2O3–. — J. Phys. Chem. A. — Vol. 103, № 8, pp. 1109—1114; 1999.
[9] A. V. Mitin. Accurate theoretical IR and Raman spectrum of Al2O2 and Al2O3 molecules. — Struct. Chem. — Vol. 22, pp. 411—418; 2011.
[10] Ch. Chang, A. B. C. Patzer, E. Sedlmayr, D. Sülzle. Ab initio studies of stationary points of the Al2O3 molecule. — Eur. Phys. J. D. — Vol. 2, pp. 57—62; 1998.
[11] A. B. Rahane, M. D. Deshpande, V. Kumar. Structural and electronic properties of (Al2O3)n clusters with n=1—10 from first principles calculations. — J. Phys. Chem. C. — Vol. 115, pp. 18111—18121; 2011.
[12] S. R. Desai, H. Wu, C. M. Rohlfing, L.-Sh. Wang. A study of the structure and bonding of small aluminum oxide clusters by photoelectron spectroscopy: AlxOy– (x=1—2, y=1—5). — J. Chem. Phys. — Vol. 106, № 4, pp. 1309—1317; 1997.
[13] P. Politzer, P. Lane, M. E. Grice. Energetics of aluminum combustion. — J. Phys. Chem. A. — Vol. 105, № 31, pp. 7473—7480; 2001.
[14] A. D. Becke. Density-functional exchange-energy approximation with correct asymptotic behavior. — Phys. Rev. A. — Vol. 38, № 6, pp. 3098—3100; 1988.
[15] Ch. Lee, W. Yang, R. G. Parr. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. — Phys. Rev. B. — Vol. 37, № 2, pp. 785—789; 1988.
[16] W. J. Hehre, L. Radom, P. v. R. Schleyer, J. A. Pople. Ab Initio Molecular Orbital Theory. — put. Chem. Vol. 7, № 3, pp. 379—383; 1986.
[17] A. A. Granovsky. Firefly Project, Moscow, Russia. Firefly (PC GAMESS) version 7.1.G, build number 5618. — http://classic. chem. msu. su/gran/firefly/index. html. e-mail: *****@***chem. msu. su.
[18] Термодинамические свойства индивидуальных веществ. Справочное издание в 4-х томах / , , и др. — 3-е изд., перераб. и расширен. — Т. III. Кн. 1. — М.: Наука, 1981. — 472 с.
[19] A. Burcat, *****scic. Third millennium ideal gas and condensed phase thermochemical database for combustion and air-pollution use with updates from active thermochemical tables. — ANL-05/20 and TAE 960 Technion-IIT. Aerospace Engineering, and Argonne National Laboratory, Chemistry Division, 2006.
