

Результаты количественного определения таблеток тропоксина методом ВЭЖХ
Номер серии | Серия 1 | Серия 2 | Серия 3 | Серия 4 | Серия 5 |
Содержание, г/табл | 0,0502 | 0,0509 | 0,0504 | 0,0502 | 0,0504 |
Метрологические характеристики (Р=95%, n=5) |
|
|
|
|
|
Определение однородности дозирования таблеток тропоксина проводили методом ВЭЖХ в условиях количественного определения. Отклонения по этому показателю всех образцов таблеток тропоксина не превышали норм установленных ГФ СССР XI издания и составляли ± 11%.
Определение показателя «Растворение» проведено на приборе «Лопастная мешалка», скорость вращения мешалки – 100 об/мин, среда растворения – вода очищенная (1000 мл), температура среды растворения 37±0,5 °С. Содержание тропоксина в растворе определяли методом УФ-СФМ при длине волны 272 нм.
За 30 мин из всех образцов таблеток в раствор переходило от 92 до 95% действующего вещества. Кинетика высвобождения тропоксина в среду растворения представлена на рисунке 2.
Таким образом, определение содержания посторонних примесей в препарате возможно проводить и методом ТСХ, и ВЭЖХ, а количественное содержание – с помощью методов УФ-СФМ и ВЭЖХ.
В качестве основного метода для определения показателей «Посторонние примеси» и «Количественное определение» был выбран более селективный метод ВЭЖХ. Определение показателя «Растворение» из-за большого количества проб, экономичнее проводить методом УФ-СФМ.
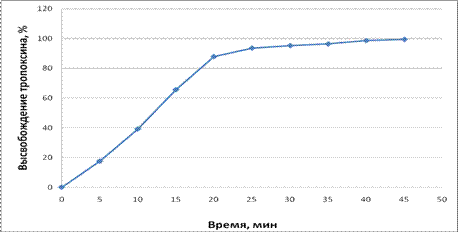
Рисунок 2. Кривая зависимости высвобождения тропоксина в раствор от времени эксперимента.
Определение подлинности таблеток тропоксина мы предложили проводить с помощью метода УФ-СФМ и ВЭЖХ.
Установление сроков годности таблеток тропоксина
Стабильность таблеток тропоксина, полученных методом прямого прессования, при хранении была изучена методом «ускоренного старения» при 60 ºС и в естественных условиях, в защищенном от света месте.
Качество таблеток практически не изменялось как при хранении в естественных условиях, так и методом «ускоренного старения» в течение срока, эквивалентного 2 годам естественного хранения. Установлен предварительный срок годности 2 года.
По результатам проведенных экспериментов установлены нормы качества твердой дозированной лекарственной формы таблеток тропоксина 50 мг (табл. 7). Установленные нормы включены в проект ФСП на таблетки тропоксина 50 мг, выполненные методом прямого прессования.
Таблица 7
Нормы качества таблеток тропоксина 50 мг
Показатели | Методы | Нормы |
Описание | Визуальный | Таблетки белого или белого со слегка кремоватым оттенком цвета плоскоцилиндрической формы без риски |
Подлинность | УФ-СФМ | УФ-спектр поглощения испытуемого раствора тропоксина в диапазоне длин волн от 250 – 350 нм должен иметь максимум поглощения при (272±2) нм |
ВЭЖХ | Времена удерживания основного пика на хроматограмме испытуемого раствора тропоксина должно соответствовать времени удерживания основного пика на хроматограмме раствора РСО тропоксина | |
Средняя масса | ГФ XI | От 0,1388 до 0,1613 г (± 7,5%) |
Распадаемость | ГФ XI | Не более 15 мин |
Однородность дозирования | ВЭЖХ | 1/10 ± 15 %; 0/10 ± 25 % |
Растворение | ОФС УФ-СФМ | Не менее 90% за 30 минут |
Посторонние примеси | ВЭЖХ | Единичной примеси – не более 0,2% Суммы примесей – не более 1,0 % |
Микробиологи-ческая чистота | ГФ XII | Категория 3А |
Количественное определение | ВЭЖХ | От 0,04625 до 0,05375 г/табл (0,05 ± 7,5 %) |
Срок годности | 2 года | |
Условия хранения | В сухом, защищенном от света месте при температуре не выше 25 °С. |
Фармацевтический анализ и стандартизация таблеток ладастена
Исследования проведены на таблетках двух составов, для влажного гранулирования и для прямого прессования, с содержанием ладастена 50 мг.
По внешнему виду таблетки, изготовленные как методом прямого прессования, так и влажным гранулированием представляли собой таблетки белого или почти белого цвета, плоскоцилиндрической формы с риской.
Средняя масса и однородность по средней массе, а так же распадаемость серийных образцов соответствовали требованиям ГФ СССР XI издания.
Хроматографические методы в анализе чистоты таблеток ладастена
Согласно схеме синтеза ладастена, технологическими примесями в его субстанции могут быть адамантан-2-он, N(2-адамантил)п-бромформанилид (АПБФА) и п-броманилин (ПБА). Примесь адамантан-2-она в лекарственной форме мы, посчитали возможным не определять, поскольку в нормативной документации на субстанцию ладастена нормируется ее отсутствие.
Определение хроматографической чистоты таблеток методом ТСХ было проведено на хроматографических пластинках Kieselgel 60 F254 (Merck) в системе гексан – ацетон (6:1). Для извлечения ладастена из лекарственных форм использовали ацетон. Детектирование проводили в УФ-свете при 254 нм, затем пластинку помещали в камеру, насыщенную парами окислов азота, а затем опрыскивали 0,5% раствором b-нафтола в спирте этиловом 95% и помещали в сушильный шкаф при температуре °С на три минуты.
На модельных смесях ладастена, технологических примесей и плацебо показано, что вспомогательные вещества не мешают извлечению и разделению ладастена и примесей, и не являются источниками дополнительных пятен на хроматограмме.
При анализе серийных образцов таблеток ладастена на линию старта наносили извлечение из таблеток в объеме, эквивалентном 200 мкг ладастена. Содержание примесей оценивали визуально, путем сравнения величины и интенсивности зон адсорбции примесей и растворов сравнения РСО ладастена, нанесенных на хроматографическую пластинку в количестве, эквивалентном 0,4 мкг (0,2%), 0,3 мкг (0,15%), 0,2 мкг (0,1%) и 0,1 мкг (0,05%) ладастена.
Для проверки пригодности хроматографической системы готовили раствор с содержанием РСО ладастена 1 мг/мл и образца примеси ПБА 0,2 мг/мл. Пригодность хроматографической системы оценивали по следующим параметрам: на хроматограмме свидетеля ладастена, нанесенного в количестве 0,1 мкг должна быть четко видна зона его адсорбции; значение Rs п-броманилина относительно ладастена должно быть 0,22±0,03.
При анализе образцов таблеток ладастена двух составов при выпуске примесей обнаружено не было.
Разработку методики ВЭЖХ проводили на стальной колонке Ultra 5 мкм C18 (250 x 4,6 мм) с предколонкой Ultra 5 мкм C18 (30 x 4,6 мм). Хроматографическая подвижность ладастена и идентифицированных примесей была изучена в подвижных фазах, включавших ацетонитрил, метанол, воду, добавки фосфорной кислоты и 0,1М NaOH, фосфатных буферных растворов с различным значением рН. Выбор аналитической длины волны проводили с учетом совпадения максимумов областей поглощения в УФ-области спектра растворов всех исследуемых соединений.
Наилучшего разделения ладастена и примесей удалось в подвижной фазе (ПФ): ацетонитрил – метанол – вода, доведенная до рН 2,50 ± 0,05 фосфорной кислотой (7:2:2) с применением программирования скорости. Скорость потока ПФ – 1 мл/мин в течение 4 мин, затем увеличение от 1 до 1,9 мл/мин за 0,2 мин. Температура колонки комнатная, аналитическая длина волны 245 нм, концентрация испытуемого раствора 1 мг/мл, объем пробы 20 мкл.
В этих условиях относительные времена удерживания АПБФА и ПБА (относительно ладастена) составляли 0,22 и 0,24 соответственно. Пределы обнаружения составляли для АПБФА-0,002 мкг; ПБА-0,0021 мкг; ладастена-0,01 мкг. Хроматограмма серийного образца субстанции ладастена с добавлением свидетелей примесей в содержании 1% от концентрации ладастена в пробе представлена на рисунке 3.
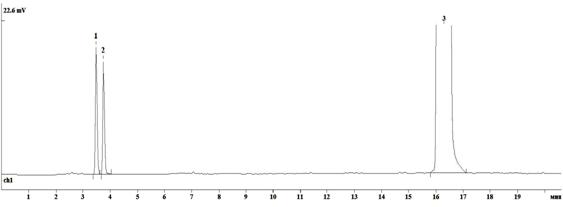
Рисунок 3. Хроматограмма раствора ладастена (1 мг/мл) и свидетелей примесей (1- АПБФА; 2- ПБА, концентрация каждого соединения 0,01 мг/мл)
Для проверки пригодности хроматографической системы готовили раствор с содержанием РСО ладастена 0,01 мг/мл и образца примеси ПБА 0,01 мг/мл. Пригодность системы оценивали по эффективности хроматографической колонки, рассчитанной по пику ладастена (не менее 5000 теоретических тарелок), коэффициенту асимметрии пика ладастена (не более 1,5), относительному стандартному отклонению, рассчитанному для площадей пиков ладастена по пяти измерениям (не более 2%) и относительному времени удерживания ПБА относительно ладастена (0,24±0,01).
Анализ извлечений из плацебо таблеток ладастена двух составов показал, что вспомогательные вещества проведению анализа не мешают.
В указанных условиях был проведен анализ серийных образцов таблеток ладастена. Оценку содержания примесей проводили относительно раствора сравнения РСО ладастена с концентрацией 0,01 мг/мл.
В образцах таблеток обоих составов были обнаружены как идентифицированные (ПБА), так и неидентифицированные примеси (tотн=0,344; 0,484; 0,556 – прямое прессование; tотн=0,344; 0,465; 0,484; 0,556 – влажное гранулирование). Содержание единичной примеси не превышало 0,2%, суммарное содержание примесей не превышало 0,5% (см. табл. 9, 10).
Разработка методов количественного анализа ладастена в таблетках
Для количественного определения ладастена в таблетках двух составов были предложены методы УФ-СФМ и ВЭЖХ.
При разработке методики УФ-СФМ извлечение ладастена из таблеточной массы проводили смесью хлороформ-спирт этиловый 95% (1:99).
Линейная зависимость оптической плотности растворов ладастена от концентрации при длине волны 260 нм (максимум поглощения растворов в УФ-области спектра), наблюдалась в интервале от 0,001 до 0,04 мг/мл (коэффициент корреляции 0,9998), рабочая концентрация 0,01 мг/мл.
Извлечение из плацебо таблеток обоих составов электромагнитное излучение при 260 нм не поглощало и не влияло на полученные результаты.
Таблица 9
Результаты определения содержания посторонних примесей
в таблетках ладастена, полученных методом прямого прессования
Обнаруженные примеси, % | Номер образца таблеток | |||
Образец 1 | Образец 2 | Образец 3 | ||
ПБА (tотн=0,235) | --- | 0,007 | 0,004 | |
Неидентифицированные примеси: | tотн=0,344 | 0,042 | 0,053 | 0,021 |
tотн=0,484 | 0,084 | 0,054 | 0,075 | |
tотн=0,556 | 0,06 | 0,04 | 0,012 | |
Сумма примесей, % | 0,186 | 0,147 | 0,112 |
Таблица 10
Результаты определения содержания посторонних примесей
в таблетках ладастена, полученных методом влажного гранулирования
Обнаруженные примеси, % | Номер образца таблеток | |||
Образец 1 | Образец 2 | Образец 3 | ||
ПБА (tотн=0,235) | 0,001 | --- | --- | |
Неидентифицированные примеси: | tотн=0,344 | 0,053 | 0,049 | 0,055 |
tотн=0,465 | --- | --- | 0,021 | |
tотн=0,484 | 0,094 | 0,093 | 0,095 | |
tотн=0,556 | 0,045 | 0,043 | 0,046 | |
Сумма примесей, % | 0,193 | 0,185 | 0,217 |
Точность и сходимость, а также диапазон применения методики оценивали путем анализа модельных смесей плацебо с известным содержанием ледестена (от 80 до 120% от рабочей концентрации ладастена). Относительная ошибка разработанной методики количественного определения содержания тропоксина, определенная на модельных смесях не превышает 1,0 % (табл. 11).
Таблица 11
Результаты количественного определения ладастена методом УФ-СФМ на модельных смесях для таблеток, полученных прямым прессованием
Взято ладастена, г | 0,0802 | 0,0852 | 0,0905 | 0,0953 | 0,1022 |
Найдено ладастена, г | 0,0800 | 0,0851 | 0,0905 | 0,0951 | 0,1012 |
Метрологические характеристики (Р=95%, n=5) |
|
|
|
|
|
Взято ладастена, г | 0,1037 | 0,1121 | 0,1163 | 0,1225 | |
Найдено ладастена, г | 0,1034 | 0,1118 | 0,1160 | 0,1213 | |
Метрологические характеристики (Р=95%, n=5) |
|
|
|
|
Количественное содержание действующего вещества во всех серийных образцах таблеток ладастена соответствовало требованиям ГФ СССР XI издания (50 мг ± 7,5%) и составляло от 4,87 до 50,13 мг/табл. (см. табл. 12, 13).

|
Из за большого объема этот материал размещен на нескольких страницах:
1 2 3 |
