
Партнерка на США и Канаду по недвижимости, выплаты в крипто
- 30% recurring commission
- Выплаты в USDT
- Вывод каждую неделю
- Комиссия до 5 лет за каждого referral
Показано, что для НМСН 1В типа более характерен ранний дебют заболевания (до 5 летнего возраста), а также большая выраженность и генерализация процесса. Это проявляется в повышении частоты встречаемости таких признаков как гипотрофия мышц голеней, мозжечковая атаксия, нарушение проприоцептивной чувствительности в ногах и руках, интенционный тремор кистей. Достоверно чаще у больных этой группы обнаруживалась поверхностная гипостезия стоп и кистей. Это связано с тем, что у больных с НМСН 1А, 1Х типа и НМСН0 отмечалась выраженная вариабельность расстройств чувствительности. Так у 16% больных с НМСН 1А, 1Х типов и НМСН0 отмечалась гиперестезия стоп, а у 7% гиперестезия кистей, которые были характерны на начальных стадиях патологического процесса, и свидетельствовали о раздражении задних рогов спинного мозга. Гиперестезия стоп отмечена нами только у 2,5.% больных с НМСН 1В типа. Кроме того, у больных с НМСН1А, 1Х типами и НМСН0 достоверно чаще отмечалась деформация стоп по типу стопы Фридрейха, в то время как у больных с НМСН 1В типа отмечались другие типы деформации стоп или их не было вовсе. Наряду с этим, показаны значимые различия в частоте встречаемости асимметрии поражения у больных с НМСН 1В типа и у больных с НМСН1А, 1Х типов и НМСН0. Этот признак обнаружен более чем у 17% больных с НМСН 1В типа и только у 1,5% больных с НМСН 1А и 1Х типами. Известно, что асимметрия поражения не характерна для наследственных нервно-мышечных заболеваний, в связи с чем, требовалось проведение дифференциальной диагностики между наследственным характером поражения и нейропатиями экзогенной и эндогенной природы. В целом можно отметить, что проведенный анализ показал наличие более тяжелой степени тяжести заболевания у больных с НМСН 1В типа.
Анализ различий между исследуемыми группами по значениям СПИ был использован непараметрический критерий Краскела-Уоллиса. Таким образом, рис. 3 отражает соотношение средних значений СПИ в исследуемых группах.
![]()
![]()
|
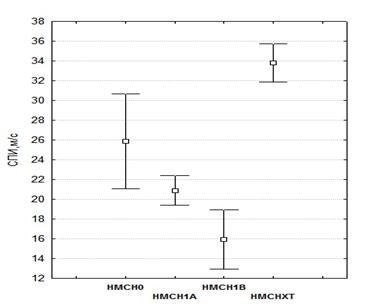
Рис. 3. Средние значения СПИ для различных типов НМСН
Таким образом, видим, что:
![]() НМСН0 и НМСН1А не обнаруживают существенных различий по значениям СПИ (соответствующие доверительные интервалы перекрываются).
НМСН0 и НМСН1А не обнаруживают существенных различий по значениям СПИ (соответствующие доверительные интервалы перекрываются).
![]() для НМСН1Х характерно существенное превышение средних значений СПИ по сравнению со всеми другими типами.
для НМСН1Х характерно существенное превышение средних значений СПИ по сравнению со всеми другими типами.
![]() НМСН1В характеризуется наименьшими средними значениями СПИ по сравнению с другими типами НМСН.
НМСН1В характеризуется наименьшими средними значениями СПИ по сравнению с другими типами НМСН.
Достоверность этого вывода подтверждается оценкой различий между исследуемыми группами НМСН на основе Критерия Краскела-Уоллиса. Результаты представлены в табл. 9.
Таблица 9. Анализ различий между исследуемыми группами пациентов по средним значениям СПИ
Средние значения СПИ, м/с | Статистика Краскела-Уоллиса | Уровень значимости | |||
НМСН0 | НМСН1А | НМСН1В | НМСН1Х | ||
25.8 ± 4.7 | 20.9 ± 1.5 | 15.9 ± 2.9 | 33.8 ± 1.9 | 84.27 | <0.001 |
Алгоритмы диагностики различных типов НМСНI
На основании результатов клинико-молекулярно-генетического анализа, проведенного нами в группе российских больных с НМСНI, разработан алгоритм их диагностики с использованием молекулярно-генетических методов. Важным этапом медико-генетического консультирования семей, отягощенных НМСН, является выявление генетического варианта с использованием молекулярно-генетических методов. Идентификация генетического варианта необходима для решения ряда проблем, основными из которых являются: определение генетического статуса родственников пробанда, определение риска рождения у них больного ребенка и планирование способов дородовой диагностики. Однако существование генетической гетерогенности и значительного сходства клинических проявлений НМСН создают значительные трудности при проведении такой диагностики с использованием дорогостоящих методов ДНК анализа. Это обусловливает необходимость создания алгоритма идентификации генетического варианта НМСН, который позволит сократить временные и материальные затраты на проведение диагностического этапа и повысит его эффективность. В основу такого алгоритма могут быть положены различия в частоте встречаемости различных вариантов НМСН, возрасте начала, типах наследования, показателях СПИ по срединному нерву и особенностях клинических проявлений и течения заболевания
Таким образом, для планирования алгоритма ДНК диагностики с целью выявления генетического варианта врачу-генетику необходимо: 1) провести генеалогический анализ; 2) определить возраст манифестации заболевания; 3) получить показатели СПИ по срединному нерву; 4) получить результаты неврологического осмотра. Диагностический алгоритм для определения типа НМСНI, адаптированный для российских пациентов, представлен на рис. 4.
![]()
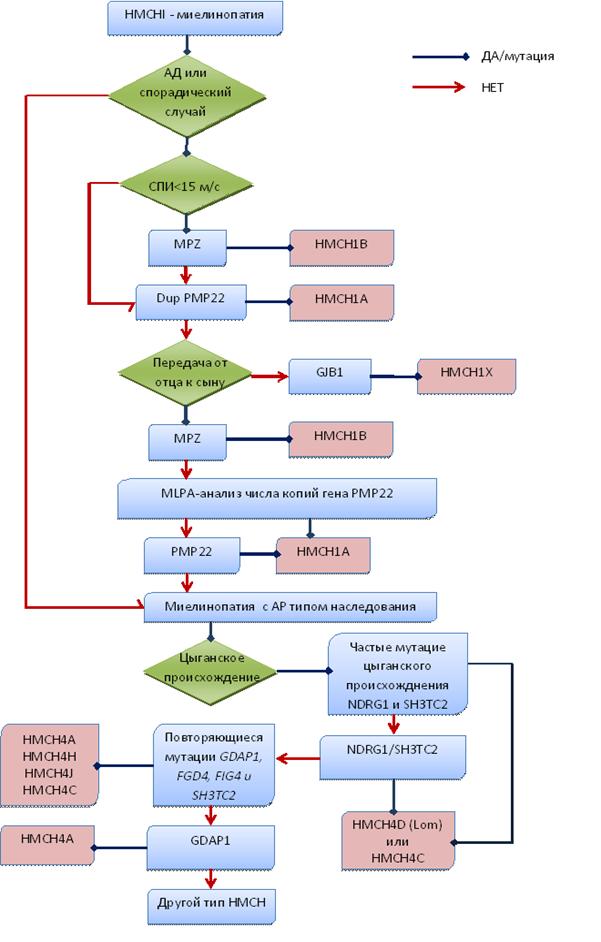
Рис. 4. Диагностический алгоритм НМСНI у российских больных
При выявлении у пробанда скоростей проведения импульса ниже 38 м/c необходимо в первую очередь исследовать мутации генов, продуктами которых являются белки миелиновой оболочки нерва.
В семьях с аутосомно-доминантным типом наследования и при наличии единственного больного в семье, важным признаком, позволяющим спланировать последовательность исследования мутаций, является СПИ по срединному нерву. При её резком снижении (<15м/с) и наличии характерных клинических признаков:
![]() Возраст дебюта до 5 лет
Возраст дебюта до 5 лет
![]() Гипостезия стоп и кистей
Гипостезия стоп и кистей
![]() Нарушение проприоцептивной чувствительности в ногах и руках
Нарушение проприоцептивной чувствительности в ногах и руках
![]() Снижение/отсутствие карпо-радиального рефлекса
Снижение/отсутствие карпо-радиального рефлекса
![]() Интенционный тремор кистей
Интенционный тремор кистей
![]() Мозжечковая атаксия
Мозжечковая атаксия
![]() Гипотрофия мышц голеней
Гипотрофия мышц голеней
![]() Асимметричность поражения
Асимметричность поражения
имеет смысл начинать поиск мутаций с гена MPZ(P0) (рис. 4).
При СПИ, колеблющихся в промежутке от 10 до 30 м/c наиболее частой причиной заболевания является дупликация на хромосоме 17р11.2-р12 в области гена PMP22, а второй по частоте причиной болезни являются наследумые Х-сцепленно доминантные мутации гена GJB1. В семьях с аутосомно-рецессивным наследованием заболевания последовательность молекулярно-генетического исследования генов построена в зависимости от частот встречаемости мутаций в различных генах при аутосомно-рецессивных формах миелинопатий: GDAP1-SH3TC2-FIG4-NDRG1.
Таким образом, нами предложен алгоритм молекулярно-генетической диагностики больных с НМСНI, в основу которого положены как результаты исследования частот встречаемости различных типов НМСНI и их клинических особенностей, так и результаты анализа спектра мутаций в генах, обусловливающих клинику данных заболеваний. Предложенный алгоритм позволяет значительно снизить экономические и временные затраты при проведении диагностики НМСН 1 типа в РФ.
ВЫВОДЫ
1. В результате поиска мутаций в генах, ответственных за возникновение доминантных вариантов НМСН 1 типа установлен вклад мутаций генов PMP22, GJB1, Р0, который составил 89% всех случаев НМСНI в РФ. В выборке из 225 больных мутации гена PMP22 явились причиной заболевания у 135 пациентов (60%). У 46 пациентов (20%) выявлены мутации гена GJB1. У 21 пациента (9%) выявлены мутации гена Р0. Мутаций генов EGR2, NEFL и LITAF не обнаружено.
Определен спектр мутаций гена GJB1 и распределение мутаций по доменам белка. Показана необходимость исследования не только кодирующей, но и некодирующей последовательности гена GJB1. Установлено, что большинство мутаций приводят к изменению трансмембранных доменов белка, ответственных за формирование стенок канала, и экстрацеллюлярной петли 2, обеспечивающей сохранность структуры канала. Показано, что реже всего поражаются высоко консервативные N - и C-домены белка коннексина 32. Определен вклад миелинопатий с АР типом наследования в общее число случаев НМСНI в РФ, который составляет 1,8%. Показано, что половина всех миелинопатий с АР типом наследования приходится на мутации в гене GDAP1. Разработаны системы детекции наиболее частых мутаций генов FGD4, FIG4, NDRG1 и SH3TC2 при демиелинизирующих нейропатиях с аутосомно-рецессивным типом наследования. В результате исключения сцепления с известными АД локусами в большой семье, где не была выявлена причина заболевания, показана дальнейшая генетическая гетерогенность демиелинизурующей формы наследственной моторно-сенсорной нейропатии. В результате анализа 34 клинических симптомов в четырех группах больных: с НМСН1А, НМСН1В, НМСН1Х типов и неустановленной формой заболевания показано отсутствие специфического симптомокомплекса, позволяющего проводить дифференциацию между НМСН1А, НМСН1Х и неустановленной формой заболевания. Выделен специфический симптомокомплекс, характерный для НМСН1В типа. Разработан алгоритм клинико-молекулярно-генетического обследования больных НМСН 1 типа, определяющий последовательность анализа генов PMP22, GJB1, MPZ, GDAP1 и часто встречающихся мутаций генов, ответственных за АР миелинопатии. Предложенный алгоритм позволяет значительно снизить экономические и временные затраты при проведении диагностики НМСН 1 типа в РФ.СПИСОК ПУБЛИКАЦИЙ ПО ТЕМЕ ДИССЕРТАЦИОННОЙ РАБОТЫ
1. , , "Исследование причин распространенности часто встречающихся мутаций гена Сх32"// Генетика человека и патология. Томск. – 2007. – С.193-194.
2. Tiburkova T. B., Schagina O. A., Dadaly E. L., Fedotov V. P., Polyakov A. V. , "Founder effect in CMT-families with mutations in GJB1 gene from different regions of Russia"// Europ. J. Hum. Genet. – 2007. - V.15. - S.1. - P.240
3. , , , , , , , , , “Спектр и частота мутаций в гене коннексина 32 (GJB1) у больных с наследственной моторно-сенсорной нейропатией 1Х типа в республике башкортостан”// Генетика. – 2008. – Т.44. - №10. – С.1385-1391.
4. T. B. Tiburkova, O. A. Schagina, E. L. Dadaly, V. P. Fedotov, A. V. Polyakov, "Hereditary motor and sensory neuropathy type I in Russia"// Europ. J. Hum. Genet. – 2008. - V.16. – S.2. - P.319.
5. , , “Исследование гена GJB1 в выборке российских больных с наследственной моторно-сенсорной нейропатией типа I” // Медицинская генетика. – 2009. - №7. - С.30-38
6. , , , “Наследственная моторно-сенсорная полинейропатия типа 4А”// Журнал неврологии и психиатрии им. . – 2010. - №5. - С.13-16.
7. , , “Классификация и алгоритмы диагностики различных генетических вариантов наследственных моторно-сенсорных полинейропатий”// Медицинская генетика. – 2011. - №4. - С.21-30.
8. , Щагина, О. А., , “Клинико-молекулярно-генетический анализ наследственной моторно-сенсорной нейропатии 1-го типа”// Материалы VI-го съезда Российского общества медицинских генетиков// Медицинская генетика. – 2010. – Т., прил. к №5. - С.178.
9. , , “Клинико-молекулярно-генетический анализ наследственной моторно-сенсорной нейропатии 1 типа” // Медицинская генетика. – 2009. - №17. - С.34-35
10. Dadaly E. L., Tiburkova T. B., Schagina O. A., Fedotov V. P. “Genotype-phenotype correlation in families with myelin protein zero (MPZ) gene mutations”// Europ. J. Hum. Genet. – 2010. - V.18. - S.1. - P.325.
11. Tiburkova T. B., Schagina O. A., Fedotov V. P., Dadaly E. L. “Molecular investigation of PMP22 gene in Russia CMT1 patients: comparison of different methods”// Europ. J. Hum. Genet. – 2010. - V.18. - S.1. - P.340.
СОКРАЩЕНИЯ
АД – Аутосомно-доминантный
АР – Аутосомно-рецессивный
ДНК – Дезоксирибонуклеиновая кислота
НМСН – Наследственная Моторно-Сенсорная Нейропатия
ПЦР – Полимеразная Цепная Реакция
СПИ – Скорость Проведения Импульса по срединному нерву
FIG4 - SAC1 lipid phosphatase domain containing
EGR2 - Early Growth Response 2 (раннееразвитиеответа 2)
FGD4 - PH domain containing 4
GDAP1 - Ganglioside-induced Differentiation-Associated Protein 1
GJB1 – Gap Junction protein, Beta 1 (белокщелевогоконтакта)
KIAA1985 (SH3TC2) - SH3 domain and TetratriCopeptide repeats 2
LITAF - Lipopolysaccharide-Induced TNF factor (липополисахарид-индуцирующий TNF фактор )
NDRG1 - N-myc Downstream Regulated Gene 1
P0 – Mielin Protein Zero (Нулевойбелокмиелина)
PMP22 – Peripheral Myelin Protein 22 (Периферический миелиновый протеин 22)

|
Из за большого объема этот материал размещен на нескольких страницах:
1 2 3 4 5 |
