
Партнерка на США и Канаду по недвижимости, выплаты в крипто
- 30% recurring commission
- Выплаты в USDT
- Вывод каждую неделю
- Комиссия до 5 лет за каждого referral

Рис. 3. Электрофореграмма ПЦР-продуктов экзона 5 гена UBE3A у пациента P23
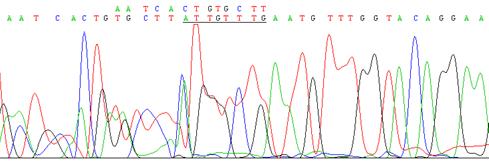

Рис. 4. Результат секвенирования участка экзона 5 гена UBE3A у пациента P23
В результате обследования родителей пациентов с СЭ, имеющих мутацию в гене UBE3A, было выявлено два случая из пяти передачи мутации от матери (пациенты D12 и C9). Оба пациента (D12 и C9) с СЭ были гетерозиготами по той же самой мутации, что и их фенотипически нормальные матери. В других трех семьях матери пациентов с СЭ не имели мутантных аллелей или признаков мозаицизма. Это означает, что мутация возникла de novo у пациента и привела к развитию заболевания.
При исследовании пациентов с СЭ зарубежными авторами, точковые мутации были обнаружены во всех кодирующих экзонах в гене UBE3A с преимущественной локализацией в экзоне 6 и в экзоне 13 (Kishino et. al., 1998; Lossie et. al., 2001; Camprubi et. al., 2009). Преобладают мутации со сдвигом рамки считывания и нонсенс мутации, которые приводят к полной потере белка или образованию белка, не способного нормально функционировать.
По литературным данным, точковые мутации в гене UBE3A, чаще обнаруживают в семейных случаях (75-80%), чем в спорадических (14 - 44%), среди пациентов с нормальным метилированием критического района (Malzac et. al., 1998; Fang et. al., 1999; Lossie et. al., 2001). В некоторых случаях, точковые мутации в гене UBE3A возникают de novo, непосредственно в материнской гамете и наследуются от матери. Предполагается, что соотношение мутаций, возникающих в женских и мужских гаметах примерно равное. Существует мнение, что замены одного нуклеотида, более вероятно, будут происходить в сперматогенезе, в результате увеличения числа клеточных делений в процессе мужского гаметогенеза, в то время как делеции или инсерции могут быть связаны с событиями, происходящими в предмейотической или мейотической стадиях в женском гаметогенезе (Camprubi et. al., 2009).
В остальных случаях молчащие мутации передаются от отца к сыну, не имея фенотипического проявления, потому что ген UBE3A имеет моноаллельную материнскую экспрессию. Мутация не проявится, если она будет передана от отца-носителя к дочери, в этом случае девочка будет здорова.
Фенотипическое проявление возникнет при передаче данной мутации от дочери к ее детям, т. е. через поколение. При формировании половых клеток у такой женщины должна произойти переустановка импринта – эпигенетической маркировки, приводящей к различиям в экспрессии родительских аллелей, которая приведет к активации материнского аллеля, в результате чего мутация проявится.
В настоящем исследовании точковые мутации в гене были выявлены у 5 из 32 (16%) больных, у которых исключены нарушения метилирования критического района.
По данным литературы, мутации в гене UBE3A обнаруживают примерно в 20% случаев у пациентов с синдромом Энжельмена. Однако в разных работах частота выявления мутаций в гене UBE3A имеет значительный разброс, и варьирует от 8% до 35% для разных выборок пациентов. Такая частота мутаций у пациентов может иметь несколько объяснений. Во-первых, вероятно наличие дефектов в других, пока не установленных локусах, расположенных либо в критическом районе хромосомы 15q11-q13, либо за его пределами на других хромосомах. Известно, что в критическом районе локализован ген ATP10C, расположенный приблизительно в 200 т. п.н. дистальнее гена UBE3A, и имеющий материнскую экспрессию. Существует предположение, что этот ген тоже играет роль в формировании фенотипических проявлений СЭ. Кроме того, существуют другие гены, повреждения в которых вызывают клинический фенотип, сходный с фенотипом СЭ. Например, ген MECP2, нарушения в котором приводят к развитию синдрома Ретта. Поскольку, эти клинические синдромы имеют похожий фенотип, необходимо проводить дифференциальную диагностику СЭ и синдрома Ретта (Jedele et. al., 2007). Также необходимо дифференцировать СЭ от синдромов X-сцепленной альфа-талассемии/синдрома умственной отсталости (ATR-X) и делеции 22q13 (Van Buggenhout et. al., 2009). Во-вторых, метод SSCP-анализа имеет некоторые ограничения чувствительности, и позволяет выявлять только 80 - 90% точковых мутаций. В-третьих, значительные различия в частоте мутаций можно объяснить недостаточно четкими критериями клинического отбора больных для лабораторной диагностики.
4. Исследование структуры и частоты молекулярной патологии, приводящей к развитию СПВ и СЭ
В результате работы получены данные о молекулярных причинах, приводящих к развитию СПВ и СЭ, на исследуемой выборке пациентов.


| - пациенты с нарушением метилирования |
| - пациенты без нарушения метилирования |
| - пациенты с делецией района отцовской хромосомы 15q11- q13 |
| - пациенты с материнской ОРД |
Рис. 5. Структура выявленной молекулярной патологии в исследуемой группе пациентов с СПВ

| - пациенты с нарушением метилирования |
| - пациенты с делецией района материнской хромосомы 15q11- q13 |
| - пациенты с отцовской ОРД |
| - пациенты без нарушения метилирования |
| - пациенты с мутациями в гене UBE3A |
| - пациенты без выявленных мутаций |
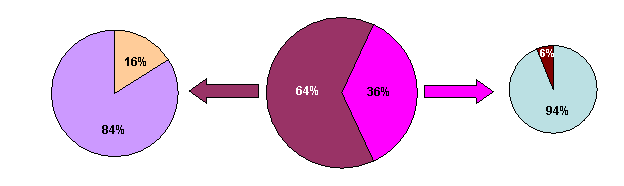
Рис. 6. Структура выявленной молекулярной патологии в исследуемой группе пациентов с СЭ
Среди пациентов с СПВ и нарушением метилирования критического района, делеция района отцовской хромосомы 15q11-q13 обнаружена в 75% случаев, а однородительская материнская дисомия в 25% случаев. У пациентов с СЭ и нарушением метилирования критического района, делеция района материнской хромосомы 15q11-q13 выявлена в 94% случаев, что несколько превышает частоту делеций, установленных в исследованиях других авторов, которая составляет 70-75% (Van Buggenhout et. al., 2009). Частота однородительской отцовской дисомии, обнаруженная у больных с СЭ в настоящем исследовании, составляет 6%. Мутации в гене UBE3A у пациентов СЭ с нормальным статусом метилирования критического района обнаружены в 16% случаев. Частота молекулярной патологии, делеций, однородительских дисомий и мутаций в гене UBE3A, сходна с частотой определяемой в других исследованиях для СПВ и СЭ. К сожалению, нам не удалось обнаружить пациентов с нарушением центра импринтинга, однако, предложенные методы исследования молекулярно-генетических причин, приводящих к СПВ и СЭ, позволяют выявить таких больных.
Полученные результаты согласуются с данными литературы, где наиболее частой причиной возникновения СПВ и СЭ становится интерстициальная делеция критического района 15q11-q13, а также однородительская дисомия. Около 20% случаев, причиной, приводящей к развитию СЭ, становятся мутации в гене UBE3A (Sartori et al., 2008).
5. Разработка ДНК - диагностического протокола для СПВ
Практическая целесообразность молекулярной диагностики СПВ и СЭ с привлечением молекулярно-генетических методов объясняется, во-первых, вариабельностью фенотипических проявлений и трудностями дифференциальной диагностики с другими наследственными патологиями; во-вторых, необходимостью профилактики в связи с исключительной тяжестью неврологических расстройств (особенно при СЭ); в-третьих, довольно высокой распространенностью в популяции и, наконец, особой необходимостью своевременного выявления наследуемых форм СПВ и СЭ, когда медико-генетическое консультирование и пренатальная диагностика могут способствовать рождению здорового потомства.
Для наиболее полного обследования пациентов с клиническим диагнозом СПВ и членов их семей предложены следующие диагностические подходы для выявления структурных и функциональных нарушений критического района хромосомы 15q11-q13 (рис. 7).
1) Цитогенетическое исследование кариотипа пациентов с СПВ с целью исключения хромосомных перестроек, представленных инвертированными дупликациями, инверсиями, сбалансированными и несбалансированными транслокациями хромосомы 15q11-q13, а также для исключения других хромосомных перестроек, приводящих к развитию сходной клинической картины. При выявлении хромосомной аномалии у пациентов с СПВ необходимо обследование родителей для исключения носительства сбалансированных хромосомных перестроек.

|
Из за большого объема этот материал размещен на нескольких страницах:
1 2 3 4 |
