

Партнерка на США и Канаду по недвижимости, выплаты в крипто
- 30% recurring commission
- Выплаты в USDT
- Вывод каждую неделю
- Комиссия до 5 лет за каждого referral
Многие химические процессы также протекают самопроизвольно, например, образование ржавчины на железе, растворение соли в воде и др. Каковы движущие силы и критерии самопроизвольных процессов?
Частицам (молекулам, атомам, ионам) присуще стремление к беспорядочному движению, поэтому система стремится перейти из более упорядоченного состояния в менее упорядоченное. Так, если, например, баллон с газом (состояние I) соединить с сосудом, то газ из баллона будет распределяться по всему объему сосуда (состояние II). При этом система из более упорядоченного состояния (с меньшим беспорядком) переходит в состояние менее упорядоченное (с большим беспорядком). Количественной мерой беспорядка системы является термодинамическая функция состояния - энтропия (S). Ее численное значение можно определить следующим образом: S = R . T. lnW, т. е. S пропорциональна lnW, где W – термодинамическая вероятность состояния системы или число вероятных микросостояний, которыми может быть реализовано данное макросостояние; W > 1.
При переходе системы из более упорядоченного состояния в менее упорядоченное (из состояния I в состояние II) энтропия системы возрастает, т. е. DS = S2 – S1 > 0.
Переход из менее упорядоченного состояния в более упорядоченное ( из состояния II в состояние I) без воздействия извне невозможен. Такой процесс называется несамопроизвольным. Понятно, что в рассматриваемом примере представляется невероятным, чтобы газ сам собой собрался в баллоне. Очевидно, что в этом случае энтропия системы уменьшается (DS = S2 – S1 < 0). Т. е.:
· все процессы, протекающие с уменьшением порядка в расположении частиц, сопровождаются увеличением энтропии, являются самопроизвольными процессами (процессы растворения, плавления, испарения, нагревания);
· все процессы, протекающие с увеличением порядка в расположении частиц, сопровождаются уменьшением энтропии, являются несамопроизвольными процессами (процессы конденсации, кристаллизации, охлаждения).
Таким образом, в изолированной системе самопроизвольные процессы протекают в сторону увеличения энтропии, DS > 0 (II закон термодинамики).
Системы, в которых протекают химические реакции, не являются изолированными, т. к. они сопровождаются тепловым эффектом, т. е. системы обмениваются энергией с окружающей средой. В неизолированных системах возможны процессы, в которых энтропия понижается. Например, при отводе тепла в окружающую среду расплав или стекло могут закристаллизоваться, а пар сконденсироваться (т. е. DS < 0).
В отличие от энтальпии, для любого вещества абсолютное значение энтропии можно вычислить либо определить экспериментальным путем. Энтропии веществ принято относить к стандартным условиям: Т = 298 К; Р = 101,3 КПа. Обозначают S0298 и называют стандартной энтропией (численное значение стандартной энтропии определяется по справочным изданиям). Энтропия вещества измеряется в Дж/моль. К.
Значениями энтропии веществ пользуются для определения изменения энтропии системы в результате соответствующих реакций. Например для реакции, записанной в общем виде:
аА +вВ + … = dD + eE + …
изменение энтропии выразится:
DS = (dSD +eSE + …) – (aSA + bSB + …) = S(ni ·Si)прод. - S(ni · Si)исх. (8)
Энтропия системы измеряется в Дж/К.
5.5. Свободная энергия Гиббса
Все химические реакции обычно сопровождаются изменением как энтропии, так и энтальпии. Связь между энтальпией и энтропией системы устанавливает термодинамическая функция состояния, которая называется свободной энергией Гиббса или изобарно-изотермическим потенциалом (G). Она характеризует направление и предел самопроизвольного протекания процессов в изобарно-изотермических условиях (р = const и Т = const). С энтальпией и энтропией системы свободная энергия Гиббса связана соотношением
G = H – TS. (9)
Абсолютное значение измерить невозможно, поэтому используется изменение функции в процессе протекания того или иного процесса:
DG = DH – TDS. (10)
Свободная энергия Гиббса измеряется в кДж/моль и кДж. Физический смысл свободной энергии Гиббса: свободная энергия системы, которая может быть превращена в работу. Для простых веществ свободная энергия Гиббса принимается равной нулю.
Знак изменения свободной энергии Гиббса DG и ее величина при Р = const определяют термодинамическую устойчивость системы:
· если в химическом процессе происходит снижение свободной энергии Гиббса, т. е. DG < 0, процесс может протекать самопроизвольно, или говорят: процесс термодинамически возможен;
· если продукты реакции имеют больший термодинамический потенциал, чем исходные вещества, т. е. DG > 0, процесс протекать самопроизвольно не может, или говорят: процесс термодинамически невозможен;
· если DG = 0, то реакция может протекать как в прямом, так и в обратном направлении, т. е. реакция обратима.
Следовательно, самопроизвольные процессы при Р=const идут с уменьшением свободной энергии Гиббса. Этот вывод справедлив как для изолированных, так и для открытых систем.
Изменение энергии Гиббса системы при образовании 1 моль вещества из простых веществ, устойчивых в данных условиях, называется энергией Гиббса образования вещества DGобр., измеряется в кДж/моль.
Если вещество находится в стандартных условиях, то энергия Гиббса образования называется стандартной энергией Гиббса образования вещества (DG0обр.298). Стандартная энергия Гиббса образования простого вещества, устойчивого в стандартных условиях, равна нулю. Значения DG0обр.298 веществ приводятся в справочниках.
Изменение энергии Гиббса, как и изменение энтальпии и энтропии, не зависит от пути процесса, поэтому изменение энергии Гиббса химической реакции DG равно разности между суммой энергий Гиббса образования продуктов реакции и суммой энергий Гиббса образования исходных веществ с учетом стехиометрических коэффициентов:
DG0298 = S(ni. DGi0298)пр. - S(ni. D Gi0298)исх.. (11)
5.6. Свободная энергия Гельмгольца
Направление протекания изохорных процессов (V = const и Т = const) определяется изменением свободной энергии Гельмгольца, которую называют также изохорно-изотермический потенциал (F):
DF = DU – TDS.
Знак изменения свободной энергии Гельмгольца DF и ее величина при V = const определяют термодинамическую устойчивость системы:
· если в химическом процессе происходит снижение свободной энергии Гельмгольца, т. е. D F < 0, процесс может протекать самопроизвольно, или говорят: процесс термодинамически возможен;
· если продукты реакции имеют больший термодинамический потенциал, чем исходные вещества, т. е. D F > 0, процесс протекать самопроизвольно не может, или говорят: процесс термодинамически невозможен;
· если D F = 0, то реакция может протекать как в прямом, так и в обратном направлении, т. е. реакция обратима.
Следовательно, самопроизвольные процессы при V=const идут с уменьшением свободной энергии Гельмгольца. Этот вывод справедлив как для изолированных, так и для открытых систем.
6. ХИМИЧЕСКАЯ КИНЕТИКА
6.1. Основные понятия химической кинетики
Химическая кинетика – раздел химии, изучающий скорости и механизмы химических реакций.
Различают гомогенные и гетерогенные химические реакции:
· гомогенные реакции протекают в однородной среде во всем объеме системы (это реакции в растворах, в газовой фазе);
· гетерогенные реакции протекают в неоднородной среде, на границе раздела фаз (горение твердого или жидкого вещества).
Основным понятием химической кинетики является понятие о скорости химической реакции. Под скоростью химической реакции понимается число элементарных актов взаимодействия в единицу времени в единице объема (если реакция гомогенная) или число элементарных актов взаимодействия в единицу времени на единицу поверхности раздела фаз (если реакция гетерогенная).
Скорость реакции характеризуют изменением концентрации какого-либо из исходных веществ или конечных продуктов реакции в единицу времени и выражают: для гомогенных реакций – моль/л·с (моль/м3·с и т. д.), для гетерогенных – моль/см2·с (моль/м2·с).
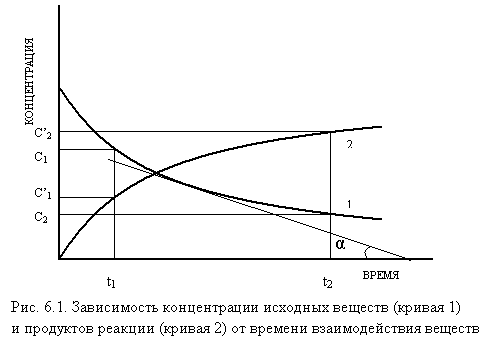 |
Различают среднюю и истинную (мгновенную) скорость реакции. Из зависимостей, представленных на рис. 6.1, следует: при химическом взаимодействии концентрация каждого из исходных веществ (кривая 1) уменьшается во времени (С2<С1, DС<0), а концентрация каждого из продуктов реакции (кривая 2) возрастает (С`2>С`1, DС>0). Следовательно, среднюю скорость (Vср) в интервале времени t1 ÷ t2 можно выразить следующим образом:
Vср =± (С2 – С1)/(t2 - t1) = ± DС/Dt. (1)
Средняя скорость является грубым приближением, т. к. в интервале времени t1 ÷ t2 она не остается постоянной. Истинная или мгновенная скорость в момент времени t (V) определяется следующим образом:
V = lim (± DС/D t) = ± dС/dt = ± С't = tg a, (2)
Dt ®0
т. е. мгновенная скорость химической реакции равна первой производной от концентрации одного из веществ по времени и определяется как tg угла наклона касательной к кривой СА = f (t) в точке, соответствующей данному моменту времени t: dС/dt = tga.
Скорость химической реакции зависит от различных факторов:
- природы реагирующих веществ;
- их концентрации;
- температуры протекания процесса;
- присутствия катализатора.
Рассмотрим более подробно влияние каждого из перечисленных факторов на скорость химической реакции.
6.2. Влияние природы реагирующих веществ
Например, взаимодействие натрия и меди с кислородом протекает в соответствии с уравнениями реакций:
1. 4Na + O2 = 2Na2O;
2. 2Cu +O2 = 2CuO.
Первый процесс протекает со скоростью V1, второй – V2, причем, V1 >> V2.
6.3. Закон действующих масс
Зависимость скорости химической реакции от концентрации реагирующих веществ определяется законом действующих масс. Этот закон установлен норвежскими учеными Гульдбергом и Вааге в 1867 г. Он формулируется следующим образом: при постоянной температуре скорость химической реакции прямо пропорциональна произведению концентраций реагирующих веществ, взятых в степенях, равных стехиометрическим коэффициентам в уравнении реакции.

|
Из за большого объема этот материал размещен на нескольких страницах:
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 |
