
Партнерка на США и Канаду по недвижимости, выплаты в крипто
- 30% recurring commission
- Выплаты в USDT
- Вывод каждую неделю
- Комиссия до 5 лет за каждого referral
Схема 21
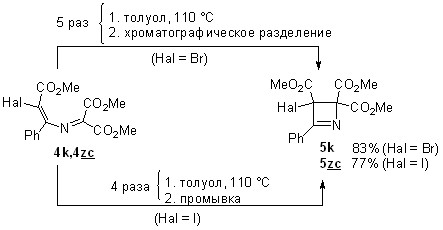
3.5. 1,5-экзо-триг-циклизация 1-ацил-4-галоген-2-азабутадиенов в 2,5-дигидрооксазолы
Поиск иных, отличных от 1,4-циклизации, путей превращения 4-ацил-4-галогензамещенных азабутадиенов решено было начать с термолиза азадиена 4za, который содержит только один акцепторный заместитель при атоме C1, что полностью исключает 1,4-циклизацию в 2,3-дигидроазет 5za (схема 22). Планируя этот эксперимент, мы не исключали возможности 1,6-циклизации этой азадиеновой системы в 2Н-1,4-оксазин 23, характерной для негалогенированных аналогов. Однако после шести часов кипячения толуольного раствора азадиена 4za даже следов оксазина 23 в реакционной смеси обнаружено не было, а вместо него методом колоночной хроматографии были выделены диастереомерные 5-метиленоксазолины 24a с общим выходом 93%. Они были разделены, и их структура была установлена с помощью стандартных спектральных методов. Для определения относительной конфигурации стереоцентров этих соединений был проведен рентгеноструктурный анализ изомера с большим значением Rf (гексан-EtOAc), установивший его rel-(R, R)-конфигурацию (рисунок 4).
Схема 22
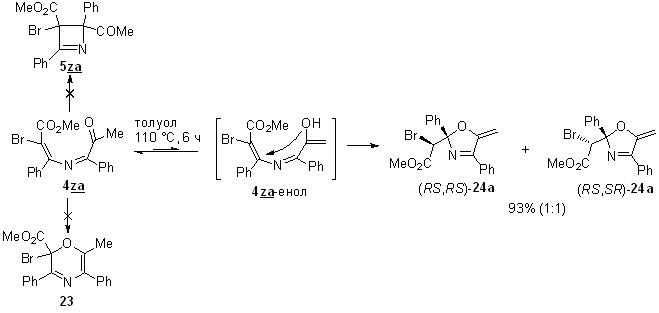
Образование 5-метиленоксазолинов 24a, вероятней всего, происходит через енолизацию азадиена и последующую 1,5-экзо-триг-циклизацию образовавшегося енола.
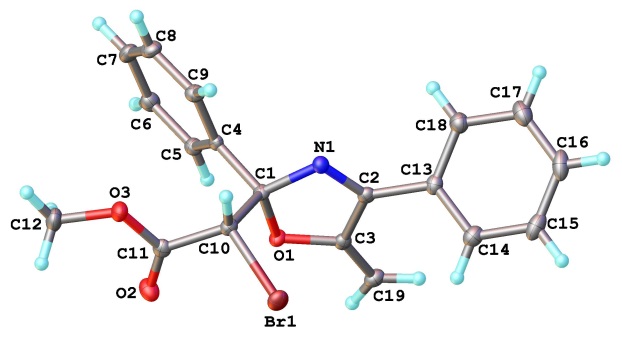
Рисунок 4. Структура соединения 24a по данным РСА
В случае азадиенов с двумя акцепторными заместителями в положении 1, в частности, тех, которые получаются из диазоацетоацетатов 3a, b или диазоацетилацетона 3i, эта термическая реакция протекает не так однозначно и приводит к сложной смеси продуктов. Однако при добавлении к растворам азадиенов 4za,4a, d,s в дихлорметане каталитического количества 1,8-диазабицикло[5.4.0]ундец-7-ена (DBU) реакция проходит при комнатной температуре за 15 минут, давая хорошие выходы 5-метиленоксазолинов 24а-d (схема 23, таблица 7). Примечательно, что хорошего выхода оксазолина 24b удалось достичь даже несмотря на гидролитическую неустойчивого азадиена 4d (таблица 7, опыт 3), который вводился в реакцию без хроматографической очистки. Z-Конфигурацию связи С=С в соединении 24d определили методом рентгеноструктурного анализа (рисунок 5).
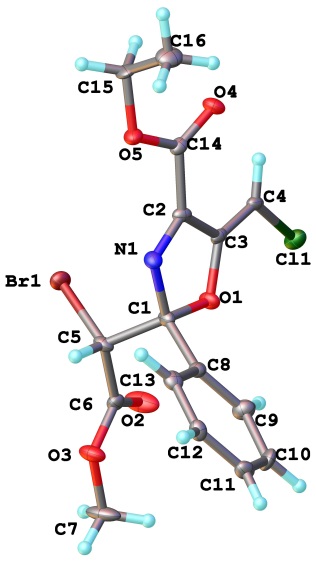
Рисунок 5. Структура соединения 24c по данным РСА
Схема 23
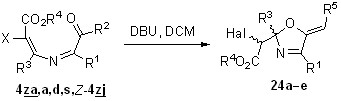
Таблица 7. DBU-катализируемая циклизация азадиенов 4za, a,d, s,Z-4zj в оксазолины 24a−e
Опыт | Азадиен 4 | X | R1 | R2 | R3 | R4 | R5 | Выход 24, % | dr (RS, RS)/(RS, SR)a |
1 | 4za | Br | Ph | Me | Ph | Me | H | 95 (a) | 1.4 : 1 |
2 | 4a | Br | CO2Et | Me | Ph | Me | H | 88 (b) | 2 : 1 |
3 | 4d | Br | CO2Et | CH2Cl | Ph | Me | Cl | 70 (c)б | 5.5 : 1 |
4 | 4s | Br | COMe | Me | Ph | Me | H | 85 (d) | 2 : 1 |
5 | Z-4zj | H | CO2Et | Me | Me | Et | H | 78 (e) |
a Диастереомерное соотношение 24 измерено методом 1H ЯМР спектроскопии.
б Выход соединения 24с в расчете на азирин 1a.
Для оценки общности этой циклизации и влияния галогена в 2-азадиене на ее протекание нами был синтезирован негалогенированный азадиен Z-4zj по известной методике [13]. Его циклизация в присутствии DBU дала негалогенированный оксазолин 24e с выходом 78%. Таким образом, эта 1,5-экзо-триг-циклизация 1-ацил-2-азабутдиенов является достаточно общей и характерной для широкого ряда енолизирующихся и устойчивых при комнатной температуре 1-ацил-замещенных 2-азадиенов.
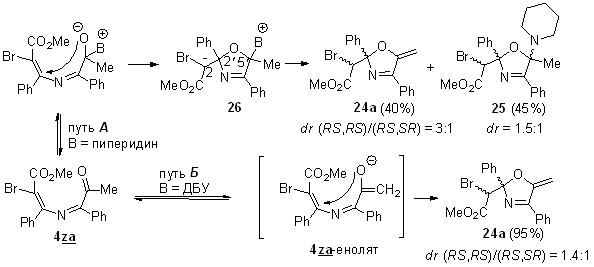
По-видимому, эта реакция может катализироваться самыми разнообразными азотистыми основаниями. Помимо DBU для каталитической циклизации азадиена 4za в пирролин 24a нами был протестирован вторичный амин, пиперидин. К нашему удивлению, наряду с целевым пирролином 24a, образовавшимся в виде смеси диастереомеров в соотношении 3:1 (40%), образовался аддукт 25, выделенный методом колоночной хроматографии с выходом 45% (схема 24). Это соединение является продуктом формального присоединения пиперидина к экзоциклической кратной связи метиленпирролина 24a. Однако отдельный эксперимент показал, что непосредственного присоединения пиперидина к 2,5-дигидрооксазолу 24а в условиях реакции не происходит. Его образование хорошо согласуется с механизмом нуклеофильного катализа реакции, ведущей к соединению 24a. Он включает присоединение на первой стадии пиперидина по карбонильной группе азадиена (схема 24) с последующей циклизацией в бетаин 26, который образуется в виде двух диастереомеров. Один из них, диастереомер (2′RS,5′RS)-26, с цис-ориентированными группами Ph и Me, претерпевают быстрый внутримолекулярный N→O прототропный сдвиг с образованием устойчивых диастереомерных аддуктов (2RS,2′RS,5′RS)-25 и (2SR,2′RS,5′RS)-25. Второй диастереомер (2′RS,5′SR)-26, с транс-ориентированными группами Ph и Me, претерпевают быстрый внутримолекулярный С→O прототропный сдвиг с отщеплением пиперидина с образованием диастереомерных метиленоксазолинов (2RS,2RS)-24a и (2RS,2′SR)-24a. На рисунке 6 представлены оптимизированные геометрии обоих диастереомеров 26 (DFT B3LYP 6-31+G(d, p), PCM для DCE), из которых видно, что межатомные расстояния между соответствующими атомами водорода и кислорода в обоих диастереомерах благоприятны для протекания упомянутых выше прототропных сдвигов.
|
|
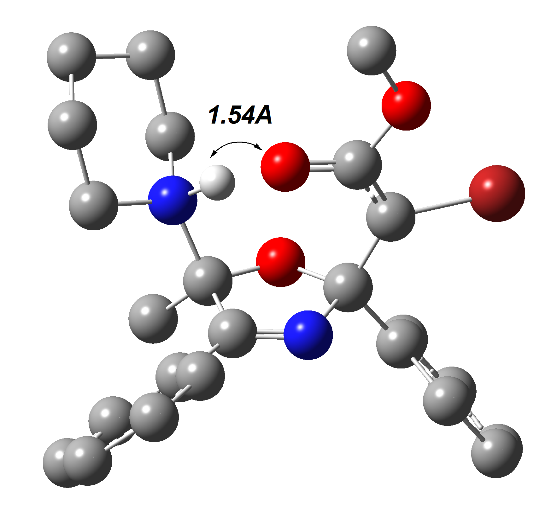
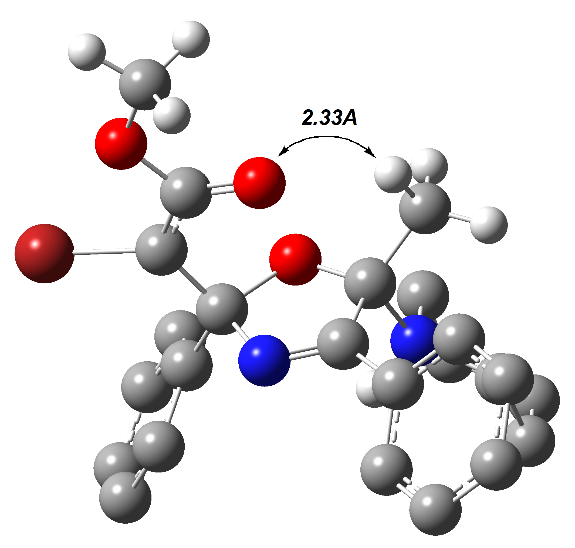
Рисунок 6. Геометрия диастереомеров 26, оптимизированных методом DFT B3LYP 31+G(d, p) (растворитель – MeOH, PCM) (для наглядности в части структурных фрагментов атомы водорода удалены)
Для реакции азадиена 4za, катализируемой DBU, мы не исключаем механизма основного катализа (схема 24, путь Б), хотя в литературе известны как реакции присоединения DBU [80], так и примеры его использования в нуклеофильном катализе [81, 82].
Схема 24
В процессе исследования 1,5-циклизации азадиенов в алкилиденоксазолины 24 было обнаружено, что заместитель при С=N связи (R1 на схеме 23) оксазолина сильно влияет на его устойчивость. В отличие от стабильного фенилзамещенного оксазолина 24a, аналоги, содержащие ацетильную или этоксикарбонильную группу в этом положении, оказались относительно неустойчивами соединениями. Так, заметная деструкция растворенных образцов в CDCl3 наблюдалась даже при -20 °C уже через несколько дней. По всей видимости, это их свойство является основной причиной неудачных экспериментов по их синтезу в условиях термолиза.
Таким образом, 1,5-экзо-триг-циклизация енолизующихся электронодефицитных 2-азабутадиенов может протекать в условиях нуклеофильного/основного катализа при комнатной температуре или в нейтральной среде при повышенных температурах. В последнем случае она конкурирует с 1,4-циклизацией в 2,3-дигидроазеты.
4. ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Температуру плавления веществ определяли на приборе SMP30, приведены неисправленные значения. ИК спектры снимали на приборе SPECORD M80 (в растворе хлороформа). Спектры ЯМР записывали на приборах Bruker DPX-300 (рабочие частоты 300 (1Н), 75 (13С) МГц) или Bruker DPX-400 (рабочие частоты 400 (1Н), 100 (13С) МГц). Масс-спектры получали на масс-спектрометре Bruker micrOTOF или MaXis. Элементный анализ выполняли на CHN-анализаторе EuroЕА3000. Данные рентгеноструктурного анализа получали на дифрактометрах Bruker SMART-6000, Agilent Gemini S-Ultra, Agilent Technologies Xcalibur и Supernova. Контроль за ходом реакции осуществляли методом ТСХ на пластинах ALUGRAM SIL G/UV254. Для разделения реакционных смесей использовали силикагель Merck 60. Толуол был перегнан над натрием, 1,2-дихлорэтан (DCE) промывали концентрированной H2SO4, затем водой, перегоняли над P2O5 и хранили над прокаленным K2CO3.
Квантово-химические расчеты были выполнены c использованием пакета программ Gaussian 09 Rev. C.01. Оптимизацию геометрии реагентов, продуктов, интермедиатов и переходных состояний проводили методом DFT mPWB1K/6-31+G(d, p) или B3LYP/6-31+G(d, p). Нахождение переходных состояний на одном энергетическом профиле с реагентами и продуктами прослеживалось внутренней координатой реакции (IRC).
Работа выполнена с использованием оборудования ресурсных центров СПбГУ «Магнитно-резонансные методы исследования», «Методы анализа состава вещества», «Вычислительный центр СПбГУ», «Рентгенодифракционные методы исследования» и «Образовательному ресурсному центру института Химии». Автор выражает благодарность сотрудникам РЦ за помощь при выполнении работы.
4.1. Синтез исходных соединений
4.1.1. Синтез метил-2-галоген-2Н-азирин-2-карбоксилатов 1а-i
Синтез (2-метокси-2-оксоэтил)трифенилфосфоний хлорида [83]
К раствору трифенилфосфина (26.2 г, 0.1 моль) в безводном этилацетате (200 мл) прибавили метил-2-хлорацетат (11.5 г, 0.106 моль). Смесь кипятили с обратным холодильником при перемешивании 30 ч. Выпавший осадок отфильтровали, промыли этилацетатом и растворили в минимальном количестве CH2Cl2. К полученному раствору по каплям добавили безводный этилацетат до полного выпадения соли фосфония, который отфильтровали и высушили в вакууме при комнатной температуре. Выход (2-метокси-2-оксоэтил)трифенилфосфоний хлорида составил 27 г (73%). Бесцветное кристаллическое вещество, т. пл. 150-153 °С (лит. т. пл. 152-153 °С [83]). Спектр ЯМР 1Н (300 МГц, CDСl3), δ, м. д.: 3.60 с (3Н, СН3), 5.83 д (2Н, СН2, J 3.4 Гц), 7.67-7.81 м (10Н, Наром), 7.89-7.96 м (5Н, Наром).

|
Из за большого объема этот материал размещен на нескольких страницах:
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 |
