


Партнерка на США и Канаду по недвижимости, выплаты в крипто
- 30% recurring commission
- Выплаты в USDT
- Вывод каждую неделю
- Комиссия до 5 лет за каждого referral
ЭВОЛЮЦИОННЫЕ АЛГОРИТМЫ ОПТИМИЗАЦИИ В ЗАДАЧЕ ПРЕДСКАЗАНИЯ ВТОРИЧНОЙ СТРУКТУРЫ БЕЛКА
1, 2
1 ГБОУ ВО МО «Университет «Дубна», Факультет естественных и инженерных наук, Дубна, Россия, *****@***com
2 Московский государственный университет имени , Факультет вычислительной математики и кибернетики, Москва, Россия, *****@***com
Предложена схема применения эволюционных алгоритмов оптимизации для предсказания вторичной структуры пептидов.
Ключевые слова: вторичная структура белка, конформационный поиск, эволюционные вычисления, глобальная оптимизация.
Введение
В работе рассматривается одна из основных задач структурной биоинформатики – предсказание трёхмерной структуры белка по аминокислотной последовательности. Белки являются макромолекулами состоящими из б-аминокислот, соединённых в цепочку пептидной связью, тем самым образуя полипептидную цепь. Предсказание структуры белка – предсказание по аминокислотной последовательности трёхмерной структуры белка, которая определяет нативное, т. е. функционально активное, состояние (выделяют вторичную, третичную и четвертичную). Короткие белки называют пептидами. В настоящей работе рассматривается задача поиска двух основных регулярных вторичных структур: б-спирали и в-листа.
Наиболее широко принимаемая гипотеза, объясняющая процесс самоорганизации белковых молекул была сформулирована Анфинсеном [1]. Основные идеи предложенной им «термодинамической гипотезы» следующие: нативное состояние белка уникально; нативное состояние белка находится в глобальном минимуме свободной энергии. Таким образом, процесс сворачивания полипептидной цепи можно представить как процесс минимизации свободной энергии белка, тогда задача предсказывания структуры сводится к задаче глобальной оптимизации.
Настоящая работа посвящена вопросу применимости стохастических эволюционных алгоритмов оптимизации к вышеописанной задаче и описанию подхода к изменению одного из параметров силового поля в процессе поиска оптимальной структуры пептида.
Численное исследование
В численных экспериментах использовалось силовое поле ROSETTA [2] в котором, при вычислении энергии пептида, сочетаются неявный растворитель, различные потенциалы и статистически полученные дынные.
В настоящей работе рассматривалось два модельных пептида длинной 10 аминокислотных остатков: A10 [3] (б-спираль), V4GGV4 [4] (в-лист). Задача поиска оптимальной структуры ставилась в непрерывном пространстве: торсионных углов главной цепи пептида (углы ц и ш, пространство поиска [-р, р]); торсионных углов главной цепи щ (стремится быть планарным, поэтому [р-д, р+д], где д = 0.2 рад.); основных торсионных углов для каждой боковой цепи ч1-4 (пространство поиска [-р, р]); длин ковалентных связей (д1 = 0.05 Е); валентных углов (д2 = 0.1 рад.) для каждого атома пептида; неосновных торсионных углов боковой цепи каждого атома (д3 = 0.1 рад.). Границы с д1-3 рассчитывались относительно идеализированных значений используемых в ROSETTA (аналогично методу CONCOORD [5]). Таким образом, размерность задач для б-спирали и в-листа составила 302 и 428 параметров соответственно.
Для поиска оптимальной структуры использовались два эволюционных алгоритма: адаптивная дифференциальная эволюция JADE [6] и эволюционная стратегия ESCH [7].
Численные эксперименты и результаты, представленные в [2], показывают, что наибольшее усложнение целевой функции порождает кулоновский (электростатический) потенциал. Поскольку данный потенциал (fa_elec) определяет нековалентные взаимодействия, было предложено использовать метод имитации отжига [8] для изменения соответствующего веса потенциала в процессе оптимизации. Такой подход был использован для рассматриваемых пептидов, результаты представлены на рис. 1-3. Все вычисления выполнены на кластере HybriLIT [9].
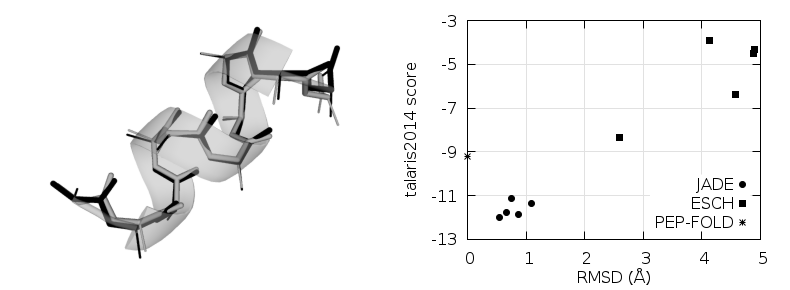
Рис. 1. Суперпозиция главных цепей пептидов, PEP-FOLD (чёрный) и JADE (серый)
Суперпозиции на рис. 1 и 2 получены с использованием 3DSS [10], оптимальные значения исследуемых структур получены с помощью сервера PEP-FOLD [11, 12], предсказывающим структуру пептида с использованием статистической информации (низкоэнергетических фрагментов). На рис. 1 и 2 показано среднеквадратичное отклонение координат атомов получаемых после оптимизации пептидов относительно найденной с помощью PEP-FOLD.
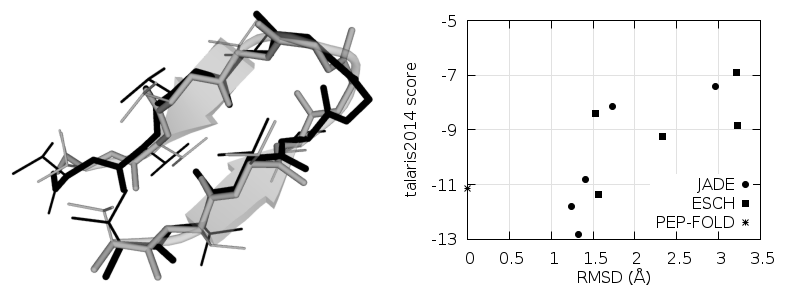
Рис. 2. Суперпозиция главных цепей пептидов, PEP-FOLD (чёрный) и JADE (серый)
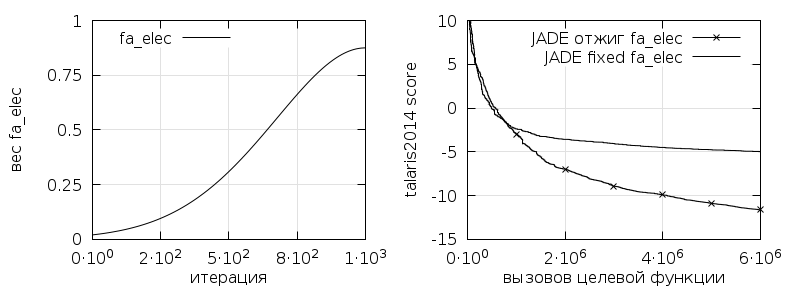
Рис. 3. Функция изменения кулоновского потенциала и сходимость для б-спирали
Выводы
На основании проведённых исследований можно заключить, что с использованием предложенной схемы эволюционные алгоритмы оптимизации способны находить оптимальную структуру коротких модельных пептидов в полноатомном разрешении. Целью дальнейшей работы является расширение схемы разбиения весов силового поля, пополнение исследуемых пептидов реальными, а также применение эволюционных алгоритмов в задаче поиска оптимального положения пептида на белке.
Литература
1. Anfinsen C. Principles that Govern the Folding of Protein Chains // Science. – Vol. 181(4096). – Jul. 1973. – Pp. 330-331.
2. O'Meara M. J., Leaver-Fay A., Tyka M. D., Stein A., Houlihan K., DiMaio F., Bradley P., Kortemme T., Baker D., Snoeyink J., Kuhlman bined Covalent-Electrostatic Model of Hydrogen Bonding Improves Structure Prediction with Rosetta // Journal of Chemical Theory and Computation. – Vol. 11(2). – 2015. – Pp. 609-622.
ng S. S. Helix Folding Simulations with Various Initial Conformations // Biophysical Journal. – Vol. 66. – Jan. 1994. – Pp. 1796-1803.
ng S. S. Monte Carlo Simulations of в-Hairpin Folding at Constant Temperature // Biophysical Journal. – Vol. 76 – Jan. 1999. – Pp. 164-175.
5. de Groot B. L., van Aalten D. M., Scheek R. M., Amadei A., Vriend G., Berendsen H. J. Prediction of protein conformational freedom from distance constraints // Proteins. – Vol. 29(2). – Oct. 1997. – Pp. 240-251.
6. Zhang J., Sanderson A. JADE: Adaptive differential evolution with optional external archive // IEEE Transactions on Evolutionary Computation. – Vol. 13(5). – 2009. – Pp. 945-958.
7. Silva-Santos C. H., Goncalves M. S., Hernandez-Figueroa H. E. Designing Novel Photonic Devices by Bio-Inspired Computing // IEEE Photonics Technology Letters. – Vol. 22(10). – 2010. – Pp. 1177-1179.
8. Kirkpatrick S., Gelatt C. D., Vecchi M. P. Optimization by Simulated Annealing // Science. – Vol. 220(4598). – 1983 – Pp. 671-680.
9. Heterogeneous Computing Cluster HybriLIT, <http://hybrilit. jinr. ru/en/>, 2015.
mathi K., Ananthalakshmi P., Roshan M. N., Sekar K. 3dSS: 3D structural superposition // Nucleic Acids Research. – Vol. 34. – Jul. 2006. – Pp. 128-132.
11. Shen Y., Maupetit J., Derreumaux P., Tuffery P. Improved PEP-FOLD approach for peptide and miniprotein structure prediction // Journal of Chemical Theory and Computation. – Vol. 10. – 2014 – Pp. 4745-4758.
12. Thevenet P., Shen Y., Maupetit J., Guyon F., Derreumaux P., Tuffery P. PEP-FOLD: an updated de novo structure prediction server for both linear and disulfide bonded cyclic peptides // Nucleic Acids Research. – Vol. 40. – 2012. – Pp. 288-293.
EVOLUTIONARY OPTIMIZATION ALGORITHMS IN protein secondary structure prediction
Poluyan S. V.1, Ershov N. M.2
1 Dubna State University, Faculty of Natural and Engineering Science, Dubna, Russia, *****@***com
2 Lomonosov Moscow State University, Faculty of Computational Mathematics and Cybernetics, Moscow, Russia, *****@***com
This paper proposes a scheme for application of evolutionary algorithms for peptide secondary structure prediction.
Кеу words: protein secondary structure, conformational search, evolutionary computation, global optimization.
