
Партнерка на США и Канаду по недвижимости, выплаты в крипто
- 30% recurring commission
- Выплаты в USDT
- Вывод каждую неделю
- Комиссия до 5 лет за каждого referral
А. А. ТУР
КВАНТОВО-ХИМИЧЕСКИЕ АСПЕКТЫ ТАУТОМЕРИИ В СЕМИХИНОННЫХ РАДИКАЛАХ
Карагандинский государственный университет им. , Караганда,
Республика Казахстан
tur. alexey. *****@***com
Спиновые зонды на основе семихинонных радикалов и метода динамической ЭПР-спектроскопии широко используются для исследования протолитических свойств соединений, обладающих как протоноакцепторными так и протонодонорными способностями, в качестве которых могут выступать различные органические основания и кислоты [1-3].
Для описания процесса водородотропии и протонного обмена с участием семихинонного радикала, применялись квантово-химические расчеты с использованием ab-initio метода в расширенном базисе 6-31g в неограниченном хартри-фоковском приближении, в качестве объекта исследований использовался простейший представитель оксирадикалов (I), полученный из этендиола. Структурная формула димера представлена на рисунке 1.
E = -454,1489 a. u. |
|
Е=-454,1055a. u. | б |
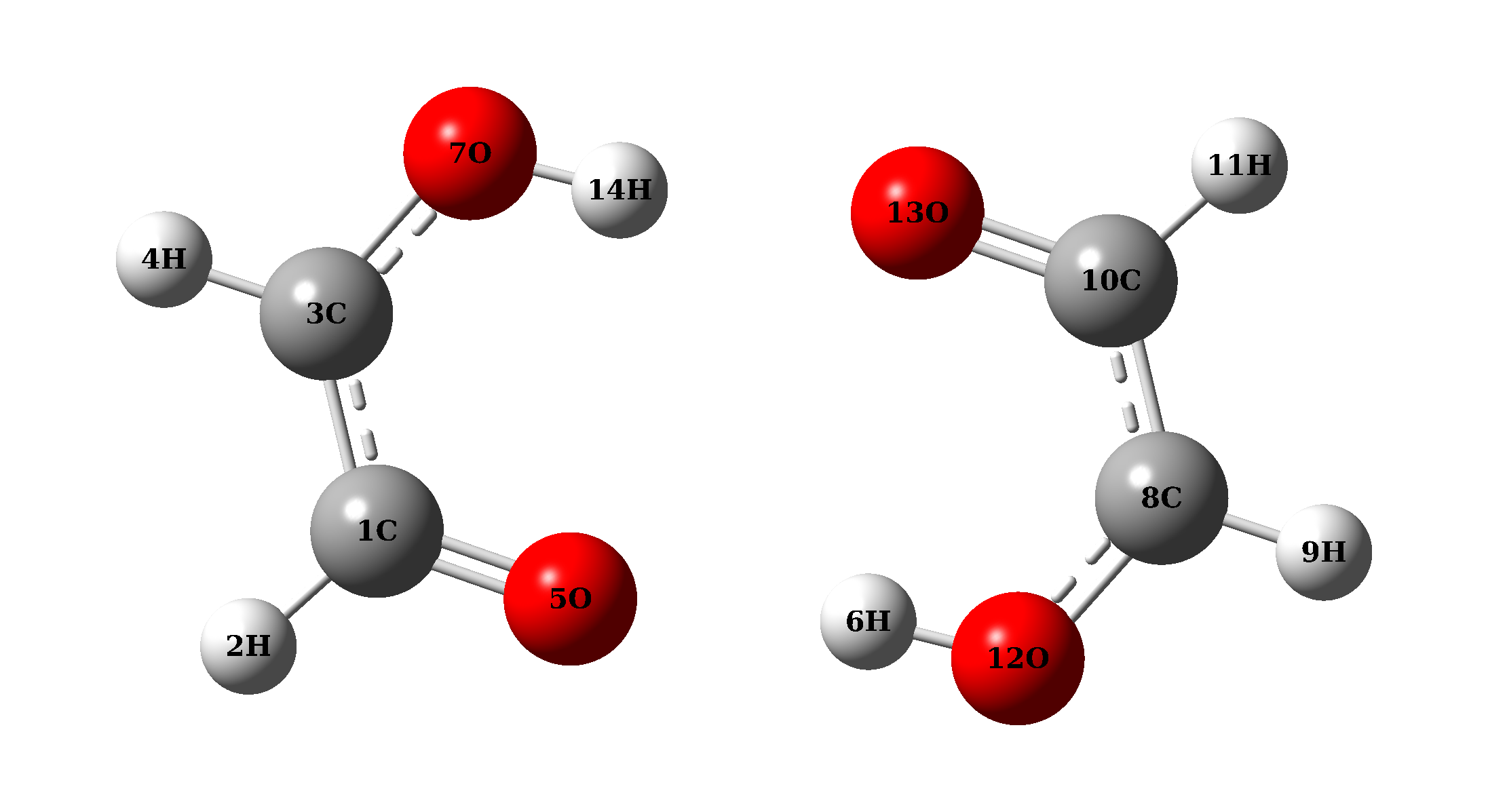
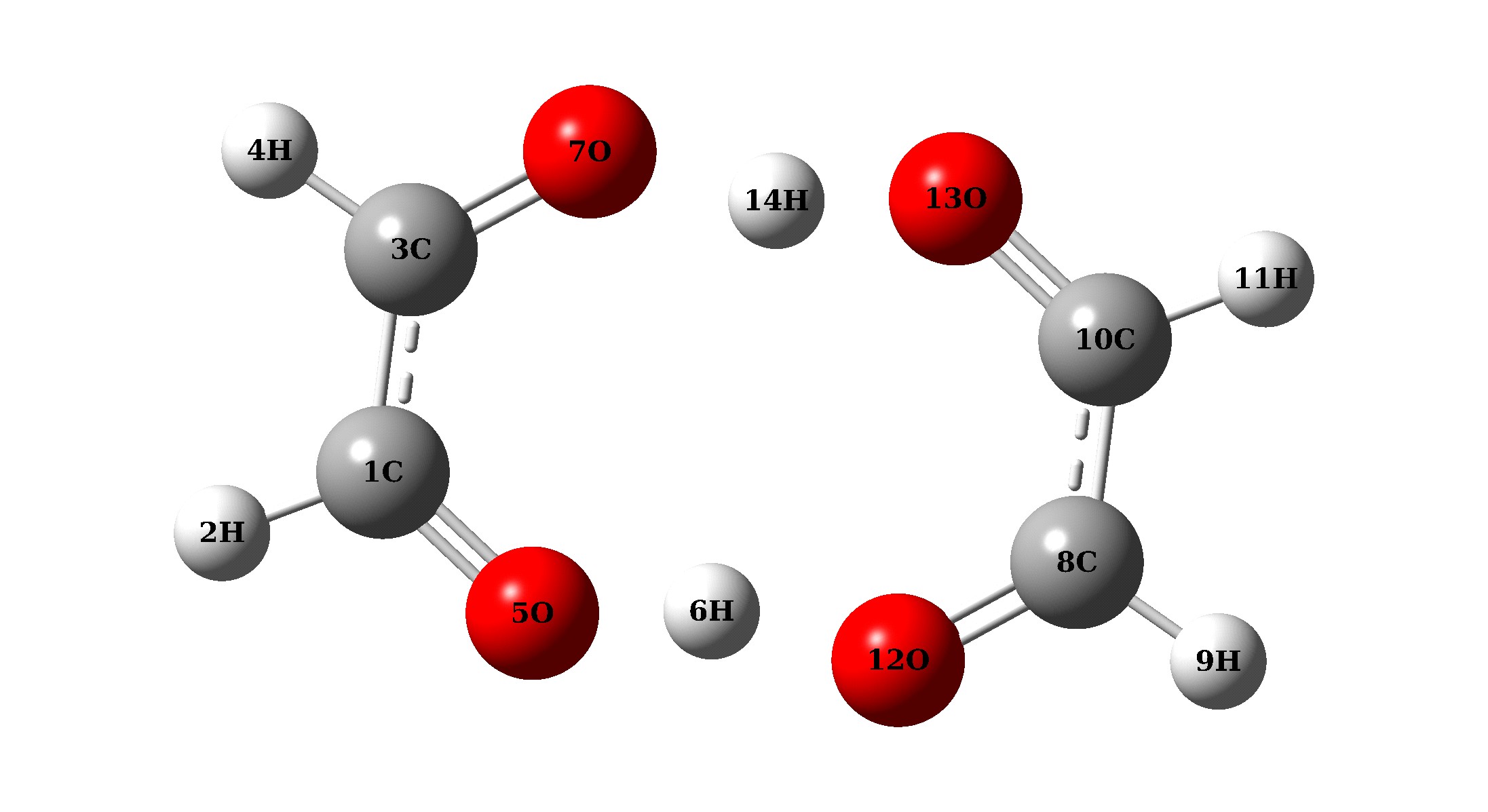

Рисунок 1 – Структура оксирадикала(I), а – межмолекулярного комплекса за счет водородной связи, б – переходного состояния
Как видно из рисунка 1, молекулы радикала ориентированы друг относительно друга таким образом, что образуют комплекс за счет водородной связи (КВС) циклического типа. А именно – водороды оксигруппы образуют водородную связь с радикальным кислородом димерной молекулы. Существование водородной связи можно установить из соотношения кинетических параметров, представленных в таблице 2:
Разность между удвоенной суммой энергии одиночной молекулы радикала этендиола и его комплекса циклического типа составляет более 44,6 кДж, что превышает значение водородной связи, соотвествующее 6,0 – 7,1 ккал.
Кроме того, на образование КВС может указывать и изменение геометрических параметров взаимодействующих молекул.
В соответствии с предложенной выше методикой - проведен квантово-химический расчет при полной оптимизации двух структур исследуемого комплекса различающихся взаимным расположением атомов водорода мигрирующих вдоль мостиков О7Н14О13 и О5Н6О12. На рисунке 1 приведены полученные таким образом начальное, конечное и переходное состояние процесса межмолекулярного протонного переноса в циклическом комплексе димера радикала этендиола:
Использование IRC процедуры при проведение квантово-химического исследования в рамках неэмпирического приближения базиса 3-21g UHF позволили определить наиболее вероятный путь, около которого проходят многие реальные траектории (рисунок 2).
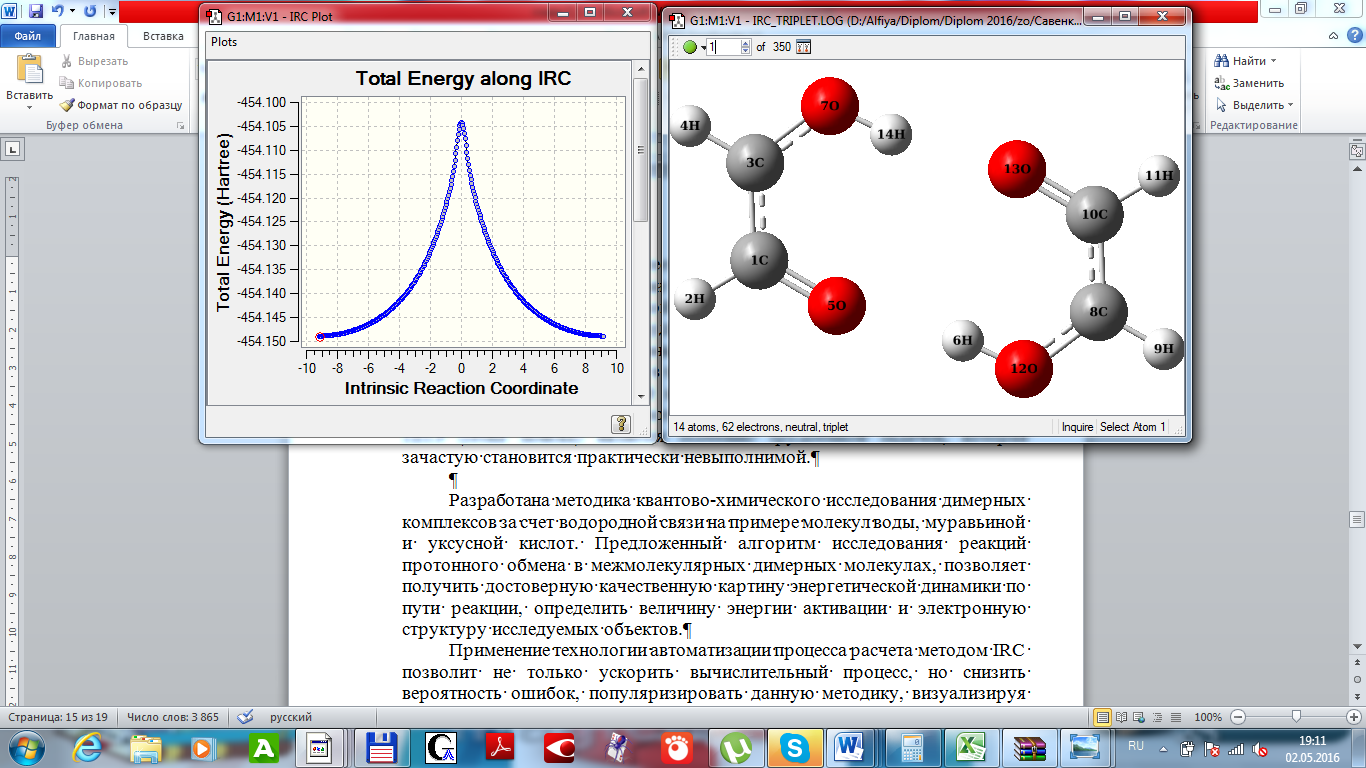
Рисунок 2 – Профиль ППЭ межмолекулярной миграции водорода в димерном комплексе радикала
В сочетании с рассчитанной геометрической структурой переходного состояния вычисления внутренней координаты реакции обеспечили эффективный метод апробирования основных областей поверхности потенциальной энергии. Это особенно важно в нашем случае, когда расчеты многомерных энергетических поверхностей необходимо проводить с использованием неэмпирических методов, и когда вычисление индивидуальных точек на ППЭ (сетка поиска) является особенно трудоемкой задачей, которая зачастую становится практически невыполнимой. Даже для построения профиля поверхности потенциальной энергии в зависимости от координат реакции в автоматическом режиме, программой исследовано 350 геометрических струк.
Представленные графические зависимости позволяют достоверно установить геометические структуры и соотвествующие им энергетические характеристики, определить активационный барьер – как разницу между исходным (конечным) состоянием и переходным состоянием: ? Е = 0,0434 a. u. = 109,68 кДж/моль
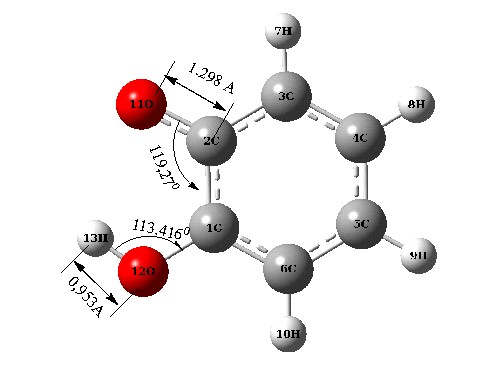
На рисунке 3 изображена молекула 2-оксифеноксила и его димер в виде комплекса образованного за счет водородной связи циклического типа.
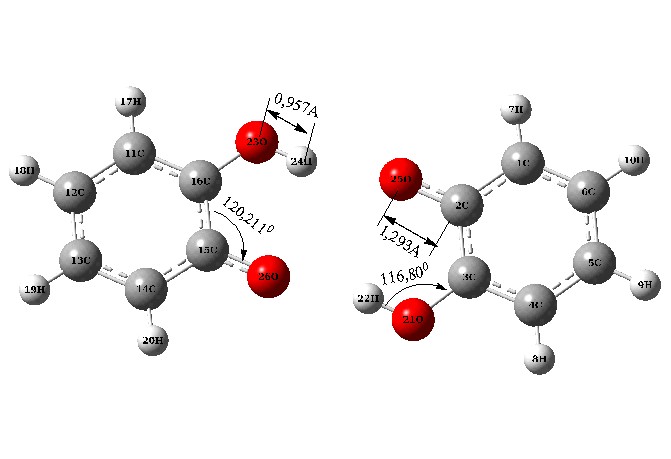
Е1 = -379,690 a. u. Е2 = -759,393 a. u.
Рисунок 3 - Структуры оксифеноксила (1) и КВС циклического типа димерной молекулы 2-оксифеноксила (2)
Как показано на рисунке 3, структура димера является является более кинетически стабильнее чем одиночная молекула оксифеноксила (Е2 - 2Е1 = 0,013 a. u.). Выигрыш в энергии, в результате образования межмолекулярных комплексов свидетельствует об энергетически более выгодном состоянии димера по сравнению с мономером.
Представленные геометрические параметры показывают, что образование димера влечет растяжение ОН связи – в гидроксильной группе, и напротив сжатие СО - связи в радикальной группировке. Кроме того, можно констатировать изменение и угловых параметров внутри пятичленного хелатного цикла *OCCOH.
Определить наиболее вероятный путь около которого проходят многие реальные траектории реакции межмолекулярного протонного протонного обмена позволит исследования IRC процедуры в рамках неэмпирического квантово-химического приближения в базисе 6-31g UHF (рисунок 4).
|
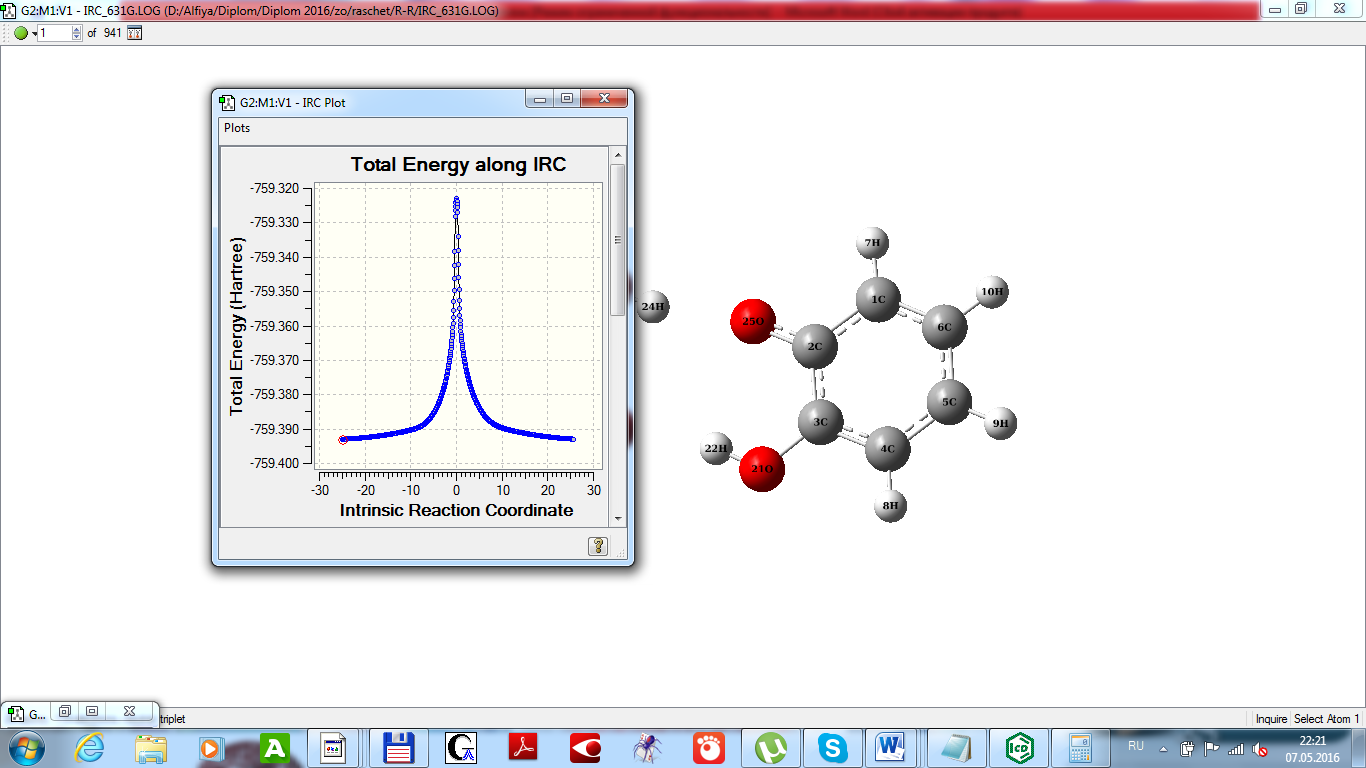
Рисунок 4 - Профиль ППЭ межмолекулярной миграции водорода в димерном комплексе 2 оксифеноксила
Наилучшее сходимость результатов и их оптимизация достигается за счет использования процедуры IRC (Intrinsic Reaction Coordinate calculation), QST2 и QST3 алгоритмы поиска переходного состояния, используется метод квадратичного синхронного транзита (Quadratic Synchronous Transit approach). Интерпретация результатов облегчается за счет автоматически сформированных графических зависимостей (Total energy along IRC, RMS gradient norm along IRC), матриц колебательных частот и наглядной визуализации молекулярных объектов.
Для интерпретации экспериментальных данных наблюдаемых в спектре ЭПР нейтрального радикала были проведены квантово-химические расчеты ортосемихинонных радикалов и их переходных состояний: незамещенного радикала, 2-оксифеноксила (RI), а также радикалов с метильным 3,6-ди-метил-2-оксифеноксила (RII) и трет. бутильным заместителями 3,6-ди-трет. бутил-2-оксифеноксила (RIII), методом UHF в базисе 3-21G, представленые на рисунках 5-6.
| а |
| б |
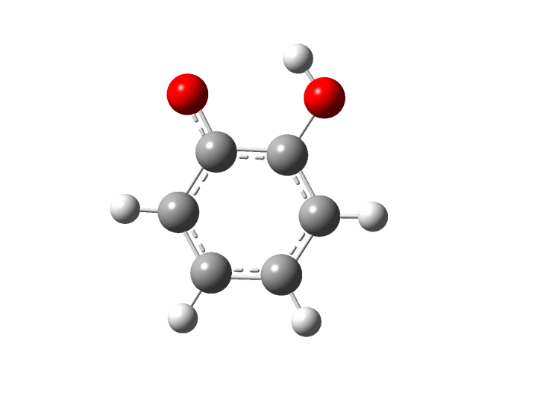
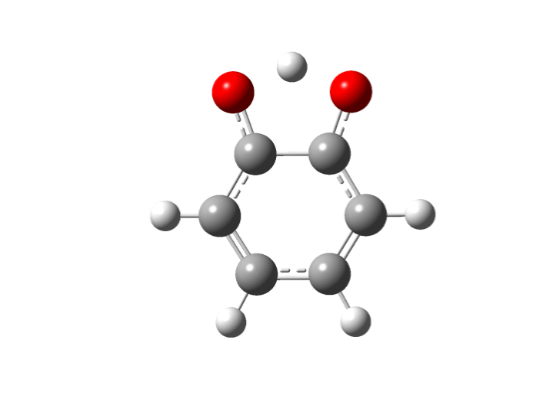
Etot = -379,69021146 a. u | Etot = -377,66495296 a. u. |
Рисунок 5 Структура незамещенного 2-оксифеноксила: а – нейтральный радикал, б – переходное состояние
| а |
| б |
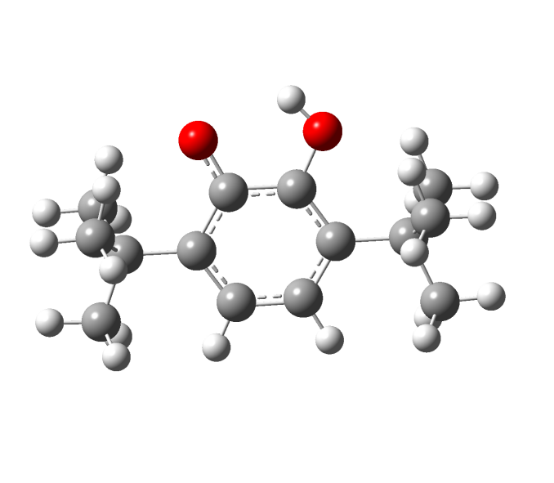
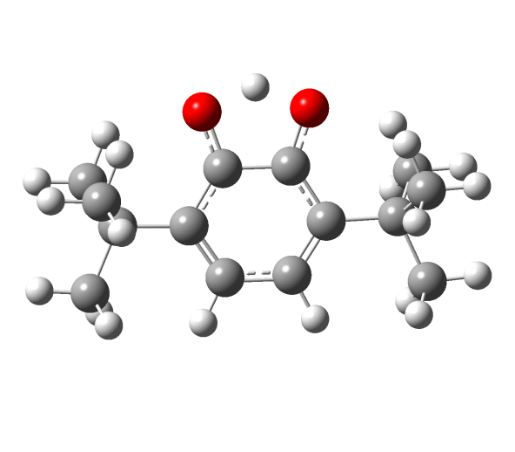
Etot = -691,83188717 a. u. | Etot = -688,23645084 a. u. |
Рисунок 6 - Структура незамещенного 3,6-ди-трет. бутил-2-оксифеноксила: а – нейтральный радикал, б – переходное состояние
Как видно из рисунков 5 - 6 структуры переходных комплексов для различных радикалов обладают рядом схожих черт, так для переходного состояния характерна более сжатая структура пятичленного комплекса ССООН, по сравнению с исходным радикальным, также удлиняется ОН-связь в комплексе по сравнению с радикалом с 0,95 до 1,25 A.
Поиск переходного состояния внутримолекулярной таутомерии, осуществлялся методом квадратичного синхронного линейного транзита. Таким образом, сначала выполняют поиск максимума в сечении ППЭ по линии, соединяющей реагенты и продукты, а затем осуществляют нахождение переходного состояния как минимума в сечении ППЭ (поверхность потенциальной энергии) по линии, перпендикулярной к линии, соединяющей реагенты и продукты и проходящей через точку максимума, найденной в приближении QST2. На рисунке 7 представлен профиль ППЭ внутримолекулярной таутомерии в 3,6-ди-трет. бутил-2-оксифеноксила.
Как показали проведенные исследования, подтверждением корректности расчета переходного состояния является наличие первой мнимой частоты колебаний для всех исследуемых структур. Сравнение энергий активации для данных радикалов показало, что без учета сольватации энергетический барьер перехода одной формы в другую составляет 1542 кДж/моль, что намного больше значений, наблюдаемых в экспериментальных спектрах ЭПР 3,6-ди-трет. бутил-2-оксифеноксила в растворе толуола порядка 12 кДж/моль [5]. Однако, полученные результаты с учетом растворителя показали величину равную 123 кДж/моль. Таким образом, полученная тенденция объясняется тем, что сольватация приводит к понижению общей энергии системы и соответственно активационного барьера для переходного состояния. Возможно, использование более расширенных базисов может привести к уменьшению данной ошибки.
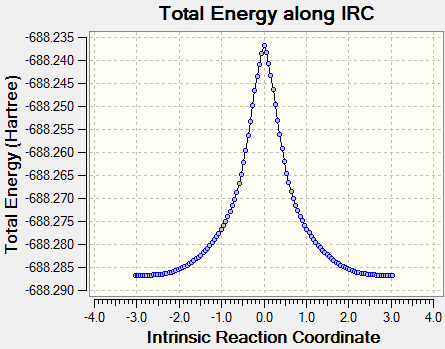
Рисунок 7 - Профиль ППЭ внутримолекулярной таутомерии в 3,6-ди-трет. бутил-2-оксифеноксиле
Отличительной способностью некоторых типов ортосемихинонных радикалов, исследованных в данной работе, является высокая внутримолекулярная подвижность в них кислого атома водорода, которая обуславливает таутомерию и двойственную протолитическую способность парамагнитных протонодоноров.
Таким образом, накоплен достаточный теоретический материал по проведению квантово-химических расчетов с использованием программы Gaussian-09, позволяющий проводить исследования внутримолекулярной миграции атомов в модельных молекулярных объектах.
Литература
