
Партнерка на США и Канаду по недвижимости, выплаты в крипто
- 30% recurring commission
- Выплаты в USDT
- Вывод каждую неделю
- Комиссия до 5 лет за каждого referral
УДК 541.515
Квантово-химические исследования магнитно-резонансных параметров спинового зонда 4-трифенилметил-6-трет. бутил-3-хлор-2-оксифеноксила
, , Чагандай I. М.
Карагандинский государственный университет имени
Семихинонные радикалы используются для оценки кинетики протолитических реакций, которые могут быть прослежены только с помощью метода ЭПР, таких, как реакции протонного переноса, обмена и электронного обмена в радикалах. Наличие в 2-оксифеноксильном радикале внутримолекулярной динамики с достаточно «высокими» скоростями должно оказывать влияние на константы СТС радикала.
Процесс таутомерии в 4-трифенилметил-6-трет. бутил-3-хлор-2-оксифеноксиле (III) можно представить в виде двухпрыжковой модели, по аналогии с семихинонным радикалом I, основное различие между которыми заключается в том, что таутомерия в радикале III является невырожденной в отличие от вырожденной таутомерии в радикале I.
| (6) |
III А III В |
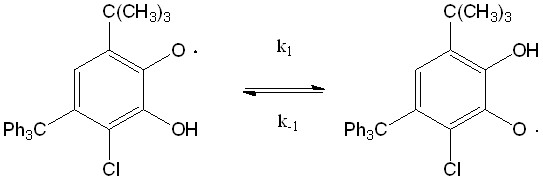
в данной схеме частицы А и В имеют различные спектральные характеристики и могут быть легко экспериментально идентифицированы.
Нами были проведены расчеты констант СТС таутомерных форм семихинонного радикала III и его переходного состояния методом Хартри-Фока. В таблице 1 представлены константы СТВ радикала полученные из спектров и рассчитанные квантово-химическими методами.
Таблица 1
Рассчитанные и экспериментальные константы СТС радикала III
Радикал | Метод | Мета- (7Н) | Пара- (7Н) | ОН (10Н) | Базисный набор | Е, a. u. |
III А | Хартри-Фок | 29.38 | -2.43 | 6-31G | -1722.006244 | |
III В | Хартри-Фок | -33.37 | -2.18 | 6-31G | -1722.006043 | |
ПС | Хартри-Фок | 11.82 | -10.30 | 6-31G | -1721.955845 | |
Хартри-Фок | 12.31 | -9.34 | 6-311G | -1722.196109 | ||
Экспер. | 0,0 | 7,8 | 1,4 |
Как видно из таблицы 1 константы СТС для протона Н(7) существенно отличаются от экспериментальных данных, однако на качественном уровне наблюдается слабая корреляция спиновой плотности на протоне бензольного кольца. Ошибка в расчетах спиновой плотности для гидроксильного протона существенно меньше для обоих структур радикала. Наблюдаемые отличия в квантово-химических расчетах по сравнению с экспериментальными данными можно связать с наличием быстрой водородотропии в радикале которая оказывает значительное влияние на локальное распределение спиновой плотности.
Для объяснения происходящих в спектрах ЭПР спинового зонда – III динамических изменений были проведены квантово-химические расчеты внутримолекулярных процессов процессов протекающих в системе, с этой целью с помощью неэмпирического приближения UHF 6-31G необходимо было выяснить строение различных форм радикала в рассматриваемой системе.
На рисунке 1 приведены различные структуры нейтральных таутомерных форм 4-трифенилметил-6-трет. бутил-3-хлор-2-оксифеноксильного радикала типа А и В. Сопоставление энергий двух структур радикала полученных для газовой фазы показывает, что структура радикала III В на 0,53 кДж/моль более устойчивее структуры III А.
|
|
|
III А | промежуточный комплекс | III В |
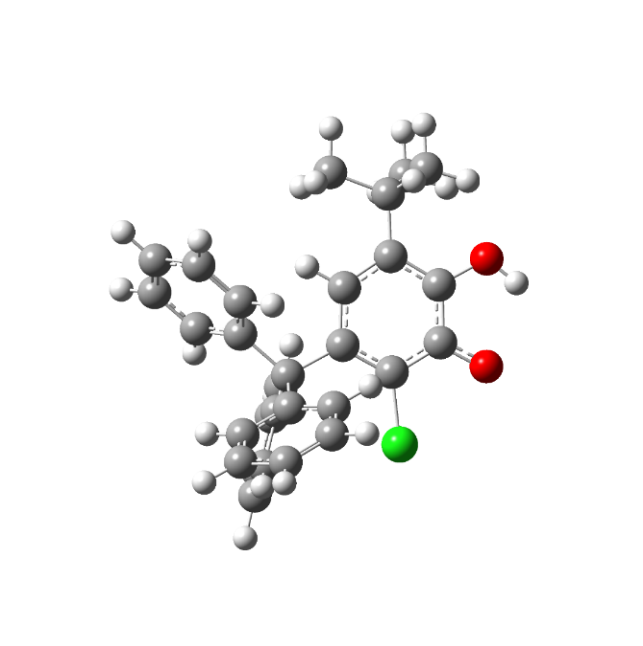
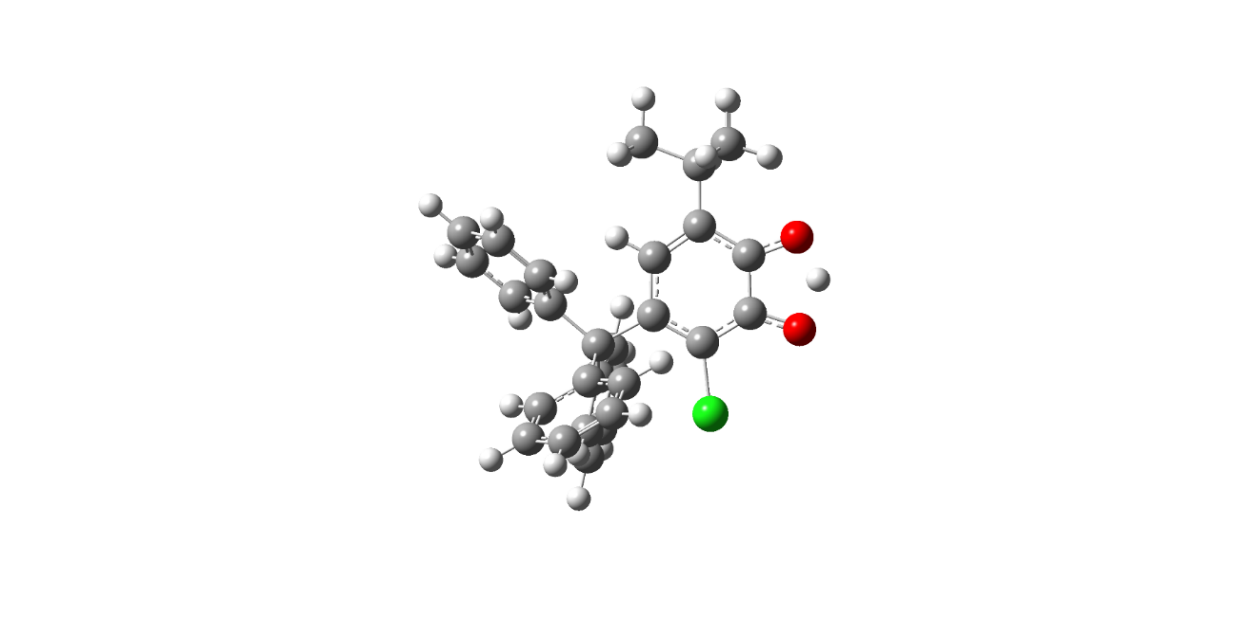
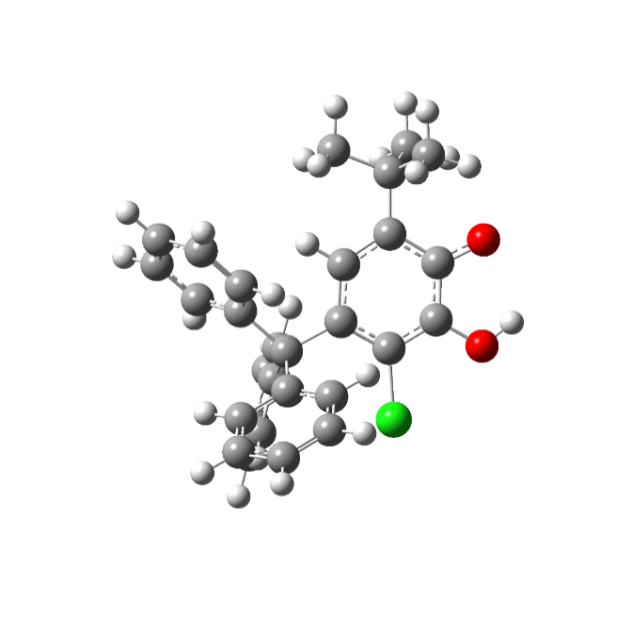
Рисунок 1. Строение таутомерных форм радикала III
Для объяснения явлений, наблюдающихся в толуольных растворах радикала нами также были рассчитаны внутримолекулярный комплекс КВС семихинонного радикала Наличие в 2-оксифеноксильном радикале внутримолекулярной динамики с достаточно «высокими» скоростями должно оказывать влияние на константы СТС радикала. С целью исследования этого влияния, то есть изменения значений констант сверхтонкого взаимодействия при увеличении длины О─Н связи, при внутримолекулярном протонном переносе была рассчитана структура промежуточного комплекса для нахождения которой использовалась процедура поиска переходного состояния.
Нами был проведен расчет пути реакции (IRC) который позволяет проследить ход реакции, приводящей к переходной структуре на ППЭ, структура промежуточного комплекса радикала III приведена на рисунке 1. На рисунке 2 представлен профиль реакции ППЭ, внутримолекулярного переноса протона, полученный методом UPM6. Как видно из рисунка 2 форма радикала III В на 2,76 кДж/моль более устойчивее, по сравнению с формой III А.
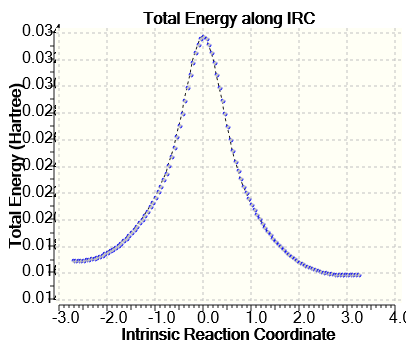
Рисунок 2. Профиль ППЭ внутримолекулярного переноса протона в радикале III
Активационные барьеры водородотропии рассчитанные методом UHF в базисе 6-31G соответственно составили для прямой реакции 132,3 кДж/моль и для обратной реакции 131,8 кДж/моль, что значительно превышает энергетические параметры, полученные из экспериментальных ЭПР-спектров радикала. Данное различие может быть обусловлено тем, что квантово-химические расчеты проводились исключительно для газовой фазы, экспериментальные параметры получены для растворов.
В работе исследованы магнитно-резонансные параметры семихинонного радикала различными методами и базисными наборами в рамках двух приближений: DFT и Хартри-Фока, и проведено сравнение расчетных параметров с экспериментальными, полученными из спектров. Квантово-химические расчеты проводились по лицензионной программе «Gaussian-2009» (США). Методами квантовой химии рассчитан профиль ППЭ реакции, внутримолекулярного переноса протона, полученный методом UPM6. Показано, что активационные параметры реакции находится в неплохом соответствии с экспериментальными данными. Различие в активационных параметрах может быть обусловлено тем, что квантово-химические расчеты проводились исключительно для газовой фазы, а экспериментальные параметры получены для растворов.
